Les récepteurs synaptiques mGluR5 en mouvement : fauteurs de troubles dans le syndrome du X fragile
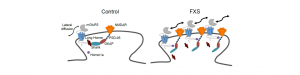
Le syndrome du X fragile (SXF) est la forme héréditaire la plus courante de déficience intellectuelle et une cause fréquente de troubles du spectre autistique (TSA). Le sous-type 5 du récepteur métabotropique au glutamate (mGluR5) est crucial dans la physiopathologie du SXF. Cependant, son dysfonctionnement au niveau subcellulaire par rapport aux phénotypes synaptiques et cognitifs du SXF est largement inexploré. mGluR5 s’associe avec un certain nombre de protéines synaptiques et ces interactions influencent fortement les propriétés dynamiques du récepteur à la synapse, ainsi que la fonction d’autres récepteurs membranaires.
Des travaux antérieurs ont démontré que l’interaction entre mGluR5 et les formes longues de la protéine d’échafaudage, Homer, est réduite chez les souris déficientes pour le gène Fmr1 (modèle murin pour SXF) et que ces altérations sont causées par une surexpression de Homer1a, l’une des isoformes d’Homer. Les conséquences de cette interaction altérée pour la dynamique des récepteurs n’avaient cependant pas été étudiées auparavant.
Dans cette étude, nous avons sondé les conséquences de la perturbation du complexe contenant mGluR5/Homer pour la mobilité de mGluR5 à la surface cellulaire, pour la fonction synaptique du récepteur N-méthyl-D-aspartate (NMDAR) et pour les phénotypes comportementaux chez les souris Fmr1 -/y.
Par le suivi d’une molécule unique, nous avons trouvé que mGluR5 était significativement plus mobile aux synapses des neurones Fmr1-/y de l’hippocampe, provoquant une augmentation de la co-localisation à la surface synaptique de mGluR5 et NMDAR. Ceci est corrélé avec une diminution de l’amplitude des courants synaptiques liée au NMDAR, à l’absence de leur dépression à long terme activée par mGluR5 et à des déficits cognitifs NMDAR / hippocampe. Ces phénomènes synaptiques et comportementaux ont été inversés en inhibant l’expression de l’isoforme courte Homer1a, dans l’hippocampe des souris Fmr1 -/y.
Notre étude fournit un important lien mécanistique entre les changements dans la dynamique de mGluR5 aux sites synaptiques et les phénotypes pathologiques du SXF. Ces découvertes sont susceptibles d’avoir des conséquences importantes pour le futur développement d’agents / stratégies thérapeutiques du SXF.
En effet, mGluR5 et NMDAR ont été proposés comme cibles thérapeutiques dans le SXF et le lien entre ces deux récepteurs devrait être pris en compte lors de la prédiction des résultats de thérapies unique ou combinée. De plus, la correction de l’équilibre altéré entre mGLluR5/Homer et mGluR5/NMDAR pourrait constituer une alternative thérapeutique prometteuse pour le développement de nouveaux agents thérapeutiques pour le traitement du SXF et des TSA.
Référence :
Aloisi E, Le Corf K, Dupuis J, Zhang P, Ginger M, Labrousse V, Spatuzza M, Georg Haberl M, Costa L, Shigemoto R, Tappe-Theodor A, Drago F, Vincenzo Piazza P, Mulle C, Groc L, Ciranna L, Catania MV, Frick A. Altered surface mGluR5 dynamics provoke synaptic NMDAR dysfunction and cognitive defects in Fmr1 knockout mice. Nat Commun. 2017 Oct 24;8(1):1103. doi: 10.1038/s41467-017-01191-2.
Contact :
Andreas Frick, Neurocentre Magendie, INSERM U1215, 146 rue Léo Saignat, 33077 Bordeaux Cedex
Courriel
Fragile X syndrome (FXS) is the most common inherited form of intellectual disability and a frequent cause of autism spectrum disorder (ASD). Metabotropic glutamate receptor subtype 5 (mGluR5) is crucially implicated in the pathophysiology of FXS. However, its dysfunction at the sub-cellular level in relation to the synaptic and cognitive phenotypes in FXS is largely unexplored. mGluR5 associates with a number of synaptic proteins and these interactions strongly influence the dynamic properties of the receptor at the synapse, as well as the function of other membrane receptors. Previous work has demonstrated that the interaction between mGluR5 and long forms of the scaffolding protein, Homer, is reduced in Fmr1 knockout mice (the mouse model for FXS) and that these alterations are caused by an overexpression of the short, activity-regulated Homer isoform, Homer 1a. The consequences of this altered interaction for receptor dynamics had not, however, been previously investigated.
In this study, we probed the consequences of the aforementioned mGluR5/Homer scaffold disruption for mGluR5 cell-surface mobility, synaptic N-methyl-D-aspartate receptor (NMDAR) function, and behavioral phenotypes in the Fmr1 knockout (KO) mouse. Using single-molecule tracking, we found that individual mGluR5 was significantly more mobile at synapses in hippocampal Fmr1KO neurons, causing an increased synaptic surface co-clustering of mGluR5 and NMDAR. This correlated with reduced amplitude of synaptic NMDAR currents, a lack of their mGluR5-activated long-term depression, and NMDAR/hippocampus dependent cognitive deficits. Crucially, these synaptic and behavioral phenomena were reversed by knocking down the short, activity-related Homer1a in the hippocampus of Fmr1 KO mice. Our study provides an important mechanistic link between changes in mGluR5 dynamics at synaptic sites and pathological phenotypes of FXS. These findings are likely to have important consequences for the future development of therapeutic agents/strategies for the treatment of FXS. In particular, both mGluR5 and NMDAR have been proposed as targets for therapeutic intervention in FXS and this altered crosstalk between these two receptors should be taken into consideration when predicting the outcome of single or combined therapies. In addition, correcting the altered balance between GluR5/Homer and mGluR5/NMDAR might provide a promising alternative for the development of novel therapeutic agents for the treatment of FXS and ASD.
Un rôle pour la sérotonine dans la motivation et l’effort !

Les neuromodulateurs modifient le traitement de l’information dans les réseaux de neurones où ils sont naturellement libérés. La sérotonine est un neuromodulateur dont le rôle est particulièrement complexe: elle est libérée dans de très nombreuses régions cérébrales, où elle interagit avec de nombreux récepteurs, et produit des effets très variés sur le comportement. Elle est par exemple impliquée dans la régulation de l’humeur, de l’anxiété, de l’impulsivité et de l’apprentissage. Il s’agit à ce titre d’une cible thérapeutique de premier plan.
La perte de motivation est un symptôme clé de la dépression mais le rôle de la sérotonine dans la régulation de la motivation reste actuellement très controversé. Des chercheurs français de l’Institut du Cerveau et de la Moelle épinière à Paris, de l’hôpital Saint Anne, associés à des collègues des Universités d’Oxford et de Manchester ont récemment publié les résultats d’une étude visant à clarifier le rôle de la sérotonine dans la régulation de la motivation.
Dans cette étude, réalisée chez des sujets sains, la motivation a été « opérationalisée » comme la capacité à produire des efforts soutenus dans lequel l’effort produit par le participant était récompensée par un gain monétaire. L’étude visait à évaluer si la sérotonine était susceptible de modifier la capacité à produire des efforts soit en exacerbant la perception du coût (fatigue, douleur, … ) soit en diminuant l’attrait des bénéfices associés à l’action. Une modification de la durée des efforts dans cette tâche pouvait donc être le résultat d’une modification de la sensibilité au coût ou au bénéfice de l’effort. Un point clé de l’étude est que les chercheurs proposent un modèle mathématique du comportement dans cette tâche qui permet de distinguer ces deux scénarios. Les résultats montrent que l’inhibition de la recapture de sérotonine améliore la performance dans cette tâche en diminuant spécifiquement le coût ressenti de l’effort : les participants recevant le traitement produisaient des efforts plus longs, et ils obtenaient ainsi des gains supérieurs à ceux recevant le placebo.
La sérotonine pourrait ainsi modifier les coûts d’une façon plus générale, qu’ils soient associés à un effort comme dans cette étude, ou à des délais ou même des punitions infligées dans de précédentes tâches de décision. Ce rôle général de la sérotonine dans le cerveau pourrait expliquer les nombreux effets déjà observés sur la régulation et la motivation du comportement.
Référence : Meyniel, F., Goodwin, G.M., Deakin, J.W., Klinge, C., MacFadyen, C., Milligan, H., Mullings, E., Pessiglione, M. and Gaillard, R., 2016. A specific role for serotonin in overcoming effort cost. Elife, 5, p.e17282.
Contact : Florent Meyniel – Chargé de recherche CEA, NeuroSpin, CEA-Saclay F-91191 Gif sur Yvette Cedex FRANCE
Un modèle animal inédit pour accélérer la lutte contre la maladie d’Alzheimer !
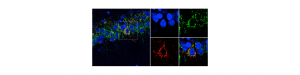
La maladie d’Alzheimer reste aujourd’hui incurable. Parmi les obstacles rencontrés pour développer des traitements efficaces figure l’impossibilité de « cibler » la maladie avant un stade avancé. Or, les caractéristiques biologiques de la maladie d’Alzheimer apparaissent au moins vingt ans avant la manifestation des symptômes cliniques (perte de mémoire, troubles émotionnels, etc.). Il faudra donc comprendre cette phase silencieuse (ou infra-clinique) pour espérer pouvoir soigner les patients alors que les atteintes cérébrales sont encore réversibles. Jusqu’à présent, il n’existait pas de modèles in vitro ou animaux qui permettent d’étudier cette longue période précédant l’apparition de symptômes cognitifs. La collaboration entre des équipes de chercheurs du département MIRCen du CEA, de l’Inserm, des universités Paris-Sud et Paris-Descartes et du CNRS a abouti au développement d’une technologie disruptive qui permet d’induire chez des rats adultes une « maladie d’Alzheimer » présentant les deux types de lésions et mimant de manière fidèle la progression de celle-ci dès ses premières phases de développement.
Dans l’article publié dans le journal Cerebral Cortex, le modèle, baptisé AgenT, est induit chez le rat à l’âge adulte par co-transfert stable des gènes humains mutés APP et PS1 à l’aide de Virus Adéno-Associés (AAV) dans l’hippocampe des animaux. Cette approche a permis la production localisée des protéines APP et PS1 dans un nombre très réduit de neurones. Ces neurones vont produire des peptides Aß42 qui vont alors diffuser dans l’ensemble du tissu hippocampique. La grande majorité du tissu hippocampique ne présente ainsi pas de modification génétique, ce qui en fait un modèle pertinent pour les formes non génétiques de la maladie qui représentent plus de 95% des cas. Sa pertinence physiopathologique a été validée en le comparant à des échantillons post-mortem de patients. La concentration des peptides Aß42 va progressivement augmenter pour atteindre en fin de vie de l’animal des concentrations comparables à celles mesurées dans l’hippocampe des patients. Une hyper-phosphorylation de la protéine Tau endogène va alors graduellement se mettre en place, simultanément les capacités de mémoire vont progressivement décliner, reproduisant la cinétique de progression de la pathologie humaine. Les plaques amyloïdes et l’angiopathie amyloïde cérébrale ne se développent qu’en fin de vie de l’animal. Des agrégats intra-neuronaux de protéine Tau hyper-phosphorylée confirment un engagement complet de la pathologie Tau.
Le fait de disposer de rongeurs modélisant de manière pertinente les stades cliniquement silencieux de la maladie va ainsi permettre 1/ d’étudier la phase précoce de la maladie, phase où le développement de celle-ci semble encore réversible, 2/ d’évaluer l’efficacité des candidats médicaments en caractérisant leurs effets sur les deux voies pathologiques (amyloïde et/ou Tau) et 3/ de définir des marqueurs sanguins spécifiques des stades précoces de la maladie, qui pourraient permettre un diagnostic dès 50 ans chez l’Homme.
Référence :
Audrain M1,2,3, Souchet B1,3,4, Alves S1,3, Fol R1,2,3, Viode A5, Haddjeri A6, Tada S1,3, Orefice NS1,3, Joséphine C3,7, Bemelmans AP3,7, Delzescaux T3,7, Déglon N8,9, Hantraye P3,7,10, Akwa Y11, Becher F5, Billard JM6, Potier B6, Dutar P6, Cartier N1,3, Braudeau J1,3.
βAPP Processing Drives Gradual Tau Pathology in an Age-Dependent Amyloid Rat Model of Alzheimer’s Disease.
Cerebral Cortex. 2017. DOI 10.1093/cercor/bhx260
1 INSERM UMR1169, Université Paris-Sud, Université Paris-Saclay, Orsay 94100, France.
2 Université Paris Descartes, Paris, France.
3 CEA, DRF, Institut Francois Jacob, MIRCen, Fontenay-aux-Roses 92265, France.
4 Université Paris Saclay, Paris, France.
5 CEA, Institut Frédéric Joliot, Service de Pharmacologie et d’Immunoanalyse, 91191, Gif-sur-Yvette, France.
6 INSERM UMR894, Centre de Psychiatrie et Neurosciences, Université Paris Descartes, Sorbonne Paris Cité, Paris, France.
7 CNRS UMR9199, Fontenay-aux-Roses 92265, Université Paris-Sud, Université Paris-Saclay, Orsay 94100, France.
8 Department of Clinical Neurosciences, Laboratory of Cellular and Molecular Neurotherapies, Lausanne University Hospital, Lausanne, Switzerland.
9 Neuroscience Research Center, Laboratory of Cellular and Molecular Neurotherapies, Lausanne University Hospital, Lausanne, Switzerland.
10 INSERM UMS27, Fontenay-aux-Roses 92265, Université Paris-Sud, Université Paris-Saclay, Orsay 94100, France.
11 INSERM U1195 and Université Paris Sud and Université Paris-Saclay, 80 rue du général Leclerc, 94276 Le Kremlin-Bicêtre, France.
Communiqué de presse :
Contact :
Jérôme Braudeau, Ph.D.
AgenT, CEA Fontenay aux Roses, Bât 61, Bureau 125, 18 route du panorama, 92260 Fontenay-Aux-Roses, France
Les bases neuronales de la phototaxis chez le poisson zèbre.
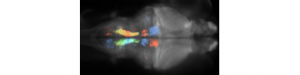
L’équipe de Georges Debrégeas au sein du Laboratoire Jean Perrin de l’Université Pierre et Marie Curie à Paris a récemment découvert les bases neuronales de la phototaxis chez le poisson zèbre. A l’état larvaire, le poisson zèbre a la capacité de naviguer vers les zones les plus lumineuses de son environnement, un comportement qu’on observe chez de nombreux organismes.
L’équipe de chercheurs a montré que l’activité d’un ensemble de neurones localisé dans le cerveau postérieur (hindbrain) du poisson zèbre appelé Hindbrain Oscillator (HBO) permet d’expliquer la phototaxis chez cet animal. En combinant des méthodes d’imagerie par nappe laser permettant d’enregistrer l’activité de l’ensemble des neurones du poisson zèbre ainsi que des techniques optogénétiques, les chercheurs ont découvert que ce HBO est une population neuronale auto-oscillante, qui contrôle la direction du regard de l’animal ainsi que l’orientation de ses mouvements de nage. L’utilisation d’un microscope à nappe laser deux photons leur a ensuite permis de découvrir que cette population neuonale est sensible à des inputs visuels de telle sorte que sa réponse dépende du contexte moteur i.e. la position des yeux. Cette réponse particulière à des stimulations visuelles biaise la navigation du poisson zèbre en direction des zones les plus lumineuses lui permettant ainsi de faire de la phototaxis
Référence
Wolf S, Dubreuil AM, Bertoni T, Böhm UL, Bormuth V, Candelier R, Karpenko S, Hildebrand DGC, Bianco IH, Monasson R, Debrégeas G. Sensorimotor computation underlying phototaxis in zebrafish. Nat Commun. 2017 Sep 21;8(1):651. doi: 10.1038/s41467-017-00310-3.
https://www-ncbi-nlm-nih-gov.gate2.inist.fr/pmc/articles/PMC5608914/
Contact
Georges Debregeas
Laboratoire Jean Perrin, UMR UPMC-CNRS 8237
4 place Jussieu, 75005 PARIS
georges.debregeas@upmc.fr
Concours « Dance your Ph.D. »

La Société des Neurosciences encourage et soutient la participation de ses membres au concours « Dance your Ph.D. », sponsorisé par la revue Science et l’AAAS, une initiative d’information sur la recherche scientifique à visée du grand public. Il s’agit d’expliquer et d’interpréter son sujet de thèse de façon ludique, dans une danse qui pourra être primée et diffusée sur internet.
Dans le cadre de l’édition 2017 de ce concours, Romain Durand-de Cuttoli, étudiant en thèse au sein de l’équipe « Neurophysiologie et comportements » dirigée par P. Faure et Alexandre Mourot (Unité Neurosciences Paris Seine) propose une vidéo : « Purple Brain: Optically disrupting the rewarding properties of nicotine ».
Vous pouvez regarder la vidéo en suivant ce lien: https://www.youtube.com/watch?v=47Men_s78_E&sns=fb
Et « liker » afin d’augmenter les chances du candidat !
Plasticité et mobilité, les clefs de la mémoire !
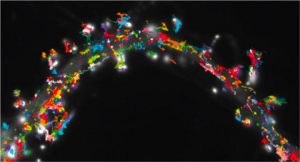
Communiqué de presse du CNRS: http://www2.cnrs.fr/presse/communique/5183.htm
Dans un effort commun, les équipes de Daniel Choquet et de Yann Humeau de l’Institut de Neuroscience Interdisciplinaire (CNRS/Université de Bordeaux) à Bordeaux et du Bordeaux Imaging Center (CNRS/Université de Bordeaux/Inserm) ont découvert récemment un mécanisme pour le stockage de l’information dans les synapses et un moyen pour contrôler ce processus de stockage.
Il y a quelques années, l’équipe de chercheurs bordelais avait découvert que les récepteurs de neurotransmetteurs n’étaient pas immobiles comme on le pensait, mais au contraire en agitation permanente. Ils avaient alors suggéré que le contrôle de cette agitation par l’activité neuronale pouvait être capable de moduler l’efficacité de la transmission synaptique en contrôlant le nombre de récepteurs présents à un instant donné dans une synapse.
Dans ces nouveaux travaux, les deux équipes sont allées plus loin dans la compréhension des mécanismes fondamentaux de stockage de l’information dans le cerveau.
Les chercheurs ont combiné des techniques de chimie, d’électrophysiologie et d’imagerie à haute résolution pour mettre au point une méthode inédite d’immobilisation des récepteurs au niveau des synapses. Grâce à cette méthode, les mouvements des récepteurs sont stoppés, ce qui permet d’étudier l’impact de leur immobilisation sur l’activité cérébrale et les capacités d’apprentissage.
Ils ont ainsi démontré que le mouvement des récepteurs est indispensable aux processus de plasticité synaptique en réponse à une activité neuronale intense. Ils ont ensuite exploré le rôle direct de la plasticité des synapses dans l’apprentissage. En apprenant à des souris à reconnaitre un environnement particulier, ils ont pu mettre en évidence que le gel du mouvement des récepteurs permet de bloquer l’acquisition de cette forme de mémoire, démontrant ainsi l’implication de la plasticité synaptique dans ce processus.
Référence:
Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Penn A.C., Zhang C.L., Georges F., Royer L., Breillat C., Hosy E., Petersen J.D., Humeau Y. and Choquet D. Nature, le 13 septembre 2017. DOI : 10.1038/nature23658
Effet de l’activité synaptique sur l’agrégation de la protéine tau: une observation clé pour la compréhension des Tauopathies
La diminution du métabolisme cérébral et les altérations de l’excitabilité synaptique sont les premiers événements associés au développement de la démence, incluant la maladie d’Alzheimer (MA) et la démence frontotemporale (DFT). Pour contrecarrer l’hypométabolisme et le dysfonctionnement des synapses, la stimulation synaptique représente une approche thérapeutique importante contre la MA. En effet, de nombreuses études ont montré que la stimulation cérébrale profonde exerce des effets bénéfiques chez les patients atteints de la MA.
Parmi les formes pathologiques de la protéine tau associées à la MA et à la DFT, les formes hyperphosphorylées et agrégées sont considérées comme les plus toxiques. Le docteur Davide Tampellini et ses collègues chercheurs au Kremlin-Bicêtre (Institut Professeur Baulieu/U 1195 Inserm), en collaboration avec une équipe nord américaine, italienne, française et espagnole, a mis en évidence les effets inverses de la stimulation cérébrale d’une part et de l’inhibition synaptique chronique d’autre part, dans des modèles in vivo et in vitro de ces tauopathies. La stimulation cérébrale profonde du cortex entorhinal de souris transgéniques de la MA (3xTg) a conduit à la réduction des formes pathologiques de tau et la protection des synapses, tandis que l’inhibition de l’activité synaptique par une déafférentation unilatérale du cortex somatosensoriel de souris transgéniques de la DFT a entraîné une accumulation massive d’oligomères de tau dans les lysosomes élargis, avec une détérioration accrue des synapses.
La stimulation synaptique a exercé une protection neuronale par un mécanisme de dégradation de la protéine tau pathologique impliquant le système autophagosome-lysosome. Notre étude a démontré que les autophagosomes étaient responsables de la livraison d’oligomères de tau aux lysosomes. En fait, le blocage de l’autophagie a réduit le nombre d’oligomères dans les somas et les lysosomes, et a accru leur accumulation dans les neurites. De plus, l’activation synaptique a augmenté la maturation de la cathepsine D et l’activité lysosomale, renforçant ainsi la dégradation de la protéine tau pathologique.
Ces données originales apportent la preuve du rôle protecteur de la stimulation synaptique contre les effets des formes pathologiques de tau dans la MA et la DFT, ainsi qu’un rationnel pour l’utilisation de thérapies telle que la stimulation cérébrale profonde pour maintenir l’activité cérébrale/synaptique.
La diminution du métabolisme cérébral et les altérations de l’excitabilité synaptique sont les premiers événements associés au développement de la démence, incluant la maladie d’Alzheimer (MA) et la démence frontotemporale (DFT). Pour contrecarrer l’hypométabolisme et le dysfonctionnement des synapses, la stimulation synaptique représente une approche thérapeutique importante contre la MA. En effet, de nombreuses études ont montré que la stimulation cérébrale profonde exerce des effets bénéfiques chez les patients atteints de la MA.
Parmi les formes pathologiques de la protéine tau associées à la MA et à la DFT, les formes hyperphosphorylées et agrégées sont considérées comme les plus toxiques. Le docteur Davide Tampellini et ses collègues chercheurs au Kremlin-Bicêtre (Institut Professeur Baulieu/U 1195 Inserm), en collaboration avec une équipe nord américaine, italienne, française et espagnole, a mis en évidence les effets inverses de la stimulation cérébrale d’une part et de l’inhibition synaptique chronique d’autre part, dans des modèles in vivo et in vitro de ces tauopathies. La stimulation cérébrale profonde du cortex entorhinal de souris transgéniques de la MA (3xTg) a conduit à la réduction des formes pathologiques de tau et la protection des synapses, tandis que l’inhibition de l’activité synaptique par une déafférentation unilatérale du cortex somatosensoriel de souris transgéniques de la DFT a entraîné une accumulation massive d’oligomères de tau dans les lysosomes élargis, avec une détérioration accrue des synapses.
La stimulation synaptique a exercé une protection neuronale par un mécanisme de dégradation de la protéine tau pathologique impliquant le système autophagosome-lysosome. Notre étude a démontré que les autophagosomes étaient responsables de la livraison d’oligomères de tau aux lysosomes. En fait, le blocage de l’autophagie a réduit le nombre d’oligomères dans les somas et les lysosomes, et a accru leur accumulation dans les neurites. De plus, l’activation synaptique a augmenté la maturation de la cathepsine D et l’activité lysosomale, renforçant ainsi la dégradation de la protéine tau pathologique.
Ces données originales apportent la preuve du rôle protecteur de la stimulation synaptique contre les effets des formes pathologiques de tau dans la MA et la DFT, ainsi qu’un rationnel pour l’utilisation de thérapies telle que la stimulation cérébrale profonde pour maintenir l’activité cérébrale/synaptique.
Référence :
Akwa Y, Gondard E, Mann A, Capetillo-Zarate E, Alberdi E, Matute C, Marty S, Vaccari T, Lozano AM, Baulieu EE, Tampellini D. Synaptic activity protects against AD and FTD-like pathology via autophagic-lysosomal degradation. Mol Psychiatry. 2017 Jul 11. doi: 10.1038/mp.2017.142
Contact :
Davide Tampellini, Ph.D.
Institut Professeur Baulieu, Bât. Gregory Pincus, 2eme étage
U 1195 Inserm – Université Paris Sud – Université Paris-Saclay
80, rue du General Leclerc
94276 Le Kremlin-Bicetre Cedex
France
Rôle de l’insuline dans le cerveau : un rôle inédit de la protéine Tau !
La protéine Tau est une des protéines majeures qui s’agrègent pour conduire à la mort neuronale dans de nombreuses maladies neurodégénératives, dont la maladie d’Alzheimer. Il s’agit d’une protéine associée aux microtubules mais qui possède bien d’autres fonctions encore méconnues. David Blum et Luc Buée de l’équipe « Alzheimer & Tauopathies », UMR-S1172 (Université de Lille CHU de Lille Inserm) viennent de démontrer qu’une fonction physiologique de la protéine Tau est de réguler les effets de l’insuline dans le cerveau. L’insuline, une hormone sécrétée par les cellules du pancréas, exerce, au niveau périphérique, une action cruciale dans le contrôle de l’homéostasie glucidique. Elle a également des actions multiples dans le cerveau en favorisant non seulement la plasticité et la mémoire mais modulant également la prise alimentaire et l’homéostasie périphérique. Les chercheurs ont démontré que les actions de l’insuline dans le cerveau étaient réduites chez des animaux ne possédant pas la protéine Tau. Ainsi, les souris déficientes en Tau présentent une réponse réduite à l’insuline au niveau de l’hippocampe, une structure du cerveau impliquée dans la mémoire. L’absence de protéine Tau provoque également des troubles du métabolisme chez les souris comme un gain anormal de poids et une intolérance au glucose, des manifestations généralement associées à l’obésité ou au diabète. Ces effets métaboliques seraient la conséquence d’une diminution des effets de l’insuline dans le cerveau, notamment sa capacité à réduire la prise alimentaire. Ce rôle de la protéine Tau est étayé par des données génétiques chez l’Homme. Cette étude fournit la première preuve d’un rôle de la protéine Tau dans les effets de l’insuline au niveau du cerveau et apporte de nouvelles pistes dans la compréhension des maladies neurodégénératives et des troubles métaboliques associés à ces pathologies.
Ces travaux ont fait l’objet d’un soutien de France Alzheimer/Fondation de France et du projet Fédératif Hospitalo-Universitaire (FHU) VasCog financé par le CHU de Lille et l’Université de Lille. L’équipe « Alzheimer & Tauopathies » appartient au LabEx DISTALZ (development of Innovative Strategies for a Transdisciplinary Approach to Alzheimer’s Disease) et au LiCEND (Lille Centre of Excellence in Neurodegenerative Disorders). Ces travaux impliquent différents laboratoires français et européens : U1011-EGID (Lille), U1016 CNRS UMR8104 (Institut Cochin, Paris), U1167-RID AGE (Lille), Laboratory of Biological Psychology, KU Leuven, Belgium, Department of Experimental Neurodegeneration, University Medical Center Goettingen, Germany
Source
Marciniak E, Leboucher A, Caron E, Ahmed T, Tailleux A, Dumont J, Issad T, Gerhardt E, Pagesy P, Vileno M, Bournonville C, Hamdane M, Bantubungi K, Lancel S, Demeyer D, Eddarkaoui S, Vallez A, Vieau D, Humez S, Faivre E, Grenier-Boley B, Outeiro TH, Staels B, Amouyel P, Balschun D, Buee L & Blum D. Tau deletion promotes brain insulin resistance (2017) The Journal of Experimental Medicine doi: 10.1084/jem.20161731.
Lien
http://jem.rupress.org/content/early/2017/06/23/jem.20161731.long
Contact chercheur
David Blum, Directeur de Recherche
UMR-S1172 « Centre de recherche Jean Pierre Aubert
« Alzheimer & Tauopathies » – LabEx DISTALZ
Faculté de Médecine-Pôle Recherche
59045 Lille France
david.blum@inserm.fr
Tel: 03 20 29 88 50
Découverte d’un nouveau mécanisme pouvant expliquer les atteintes mitochondriales dans la maladie d’Alzheimer
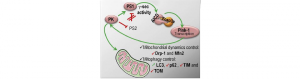
Les altérations de la dynamique mitochondriale et des processus mitophagiques sont deux des stigmates histologiques caractérisant la maladie d’Alzheimer. Peu de travaux concernent les mécanismes moléculaires qui sous-tendent ces dysfonctions. Le groupe de Cristine Alves da Costa à l’IPMC de Valbonne (Goiran et coll. Biol. Psy., 2017) a découvert que la fonction mitophagique contrôlée par PINK1 est régulée par les présénilines, ces protéines qui, quand elles sont mutées, rendent compte de la majorité des formes familiales de maladie d’Alzheimer. Ces présénilines, qui portent l’activité g-sécrétase, libèrent un fragment appelé AICD (APP Intracellular Domain) qui régule la transcription de PINK1 par un mécanisme dépendant de Foxo-3a. Ce travail est la première explication mécanistique des altérations mitophagiques dans la maladie d’Alzheimer.
De manière intéressante, il avait été établi que la parkine agit comme un facteur de transcription (da Costa et coll. Nat Cell Biol, 2009) capable de réguler la transcription des présénilines (Duplan et coll. J. Mol Cell Biol. 2013). Le présent travail montre que la parkine contrôle la fonction de PINK1 de manière totalement dépendante des présénilines. Ces résultats ont plusieurs implications. Premièrement, ils confirment la fonction transcriptionnelle de la parkine et deuxièmement, ils montrent que la parkine peut interagir avec PINK1 en amont de cette dernière, ce qui bouleverse un dogme selon lequel PINK1 interagirait toujours en amont de la parkine en contribuant à son recrutement à la mitochondrie.
Référence: Goiran T, Duplan E, Chami M, Bourgeois A, El Manaa W, Rouland L, Dunys J, Lauritzen I, You H, Stambolic V, Biféri MG, Barkats M, Pimplikar SW, Sergeant N, Colin M, Morais VA, Pardossi-Piquard R, Checler F, Alves da Costa C. β-Amyloid Precursor Protein Intracellular Domain Controls Mitochondrial Function by Modulating Phosphatase and Tensin Homolog-Induced Kinase 1 Transcription in Cells and in Alzheimer Mice Models. Biol Psychiatry. 2017 May 3. pii: S0006-3223(17)31515-9. doi: 10.1016/j.biopsych.2017.04.011. [Epub ahead of print], PMID: 28587718
Lien vers l’article : http://www.biologicalpsychiatryjournal.com/article/S0006-3223(17)31515-9/fulltext
Contact:
Dr Cristine Alves da Costa (Email : acosta@ipmc.cnrs.fr)
Institut de Pharmacologie Moléculaire et Cellulaire, Université Côte d’Azur, Centre National de la Recherche Scientifique (CNRS), Institut National de la Santé et de la Recherche Médicale (INSERM), UMR 7275, 660 Route des Lucioles, Valbonne, France,
Une avancée majeure pour la compréhension des ataxies cérébelleuses
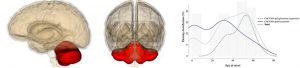
Des chercheurs de l’Institut du Cerveau et de la Moëlle Epinière (ICM) à l’Hôpital de la Pitié-Salpetrière à Paris viennent de publier une large étude de génétique épidémiologique portant sur 412 patients atteints d’ataxie cérébelleuse autosomique dominante.
L’hétérogénéité génétique de ces maladies, qui touchent le cervelet et affecte la coordination et l’équilibre et peuvent être causées par des mutations dans plus de 40 gènes, rend leur diagnostic génétique compliqué. Le plus fréquemment, la mutation incriminée est une expansion d’une répétition d’une courte séquence de nucléotides, telle qu’un triplet « CAG ». Dans de plus rares cas, on est en présence de mutations plus « conventionnelles ». Si l’épidémiologie des formes dites à expansion a été bien étudiée, ce n’est pas le cas de ces dernières.
Les chercheurs ont conçu un panel permettant de séquencer en une fois l’ensemble des 34 gènes connus pour présenter ce type de mutations dans les ataxies cérébelleuses, et l’ont utilisé chez 412 patients qui ne présentaient pas d’expansion de répétition. Cela leur a permis d’identifier une mutation responsable chez près de 15% des patients auparavant non diagnostiqués. Les gènes codant pour des canaux ioniques étaient les plus fréquemment mutés, ce qui confirme leur importance majeure dans le fonctionnement du cervelet.
Ils ont aussi comparé les présentations cliniques de l’ataxie cérébelleuse entre des patients porteurs d’une expansion et des patients présentant une mutation conventionnelle. Ces derniers commencent souvent la maladie plus tôt, mais montrent une progression plus lente et un phénotype plus pur, avec moins d’autres signes accompagnateurs.
Cette étude, en plus d’apporter un diagnostic précis à une proportion non négligeable de patients, permet donc de mieux caractériser les aspects génétiques et cliniques des ataxies cérébelleuses autosomiques dominantes, ce qui améliorera la prise en charge et le conseil génétique pour les patients.
Référence de l’article:
A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies.
Coutelier M, Coarelli G, Monin ML, Konop J, Davoine CS, Tesson C, Valter R, Anheim M, Behin A, Castelnovo G, Charles P, David A, Ewenczyk C, Fradin M, Goizet C, Hannequin D, Labauge P, Riant F, Sarda P, Sznajer Y, Tison F, Ullmann U, Van Maldergem L, Mochel F, Brice A, Stevanin G, Durr A; SPATAX network. Brain. 2017 Jun 1;140(6):1579-1594. doi: 10.1093/brain/awx081.
Lien vers l’article: https://academic.oup.com/brain/article-lookup/doi/10.1093/brain/awx081
Contact:
Dr Alexandra Durr
Reference centre for Rare diseases-Neurogenetics
ICM (Institut du Cerveau et de la Moelle épinière)
Salpêtrière Hospital – Université Pierre et Marie Curie
47 boulevard de l’Hôpital
75651 Paris CEDEX 13
alexandra.durr@icm-institute.org