Loi de programmation pour la recherche : propositions d’un collectif de sociétés savantes académiques

Collectif de Sociétés Savantes Académiques signataires : Société Française de Biologie du Développement, Société Mathématique de France, Société Française d’Ecologie et d’Evolution, Société Française d’Astronomie et d’Astrophysique, Société des Neurosciences, Société Informatique de France, Société des Professeurs d’Histoire Ancienne de l’Université, Société de Mathématiques Appliquées et Industrielles, Société des Historiens Médiévistes de l’Enseignement Supérieur Public, Société Française d’Optique, Association Française pour l’Intelligence Artificielle, Société Française de la Neutronique, Société Française des Microscopies, Comité National Français de Géographie, Association des Historiens Contemporanéistes de l’ESR, Société Française d’Exobiologie, Société Française de Statistique, Société Française de Virologie, Société Française des Sciences de l’Information et de la Communication, Société Française d’Acoustique, Société Française de Bioinformatique, Association Française de Mécanique, Association des Enseignants-Chercheurs en Psychologie des Universités, Association Française de Science Economique, Association Française de Sociologie, Société Française de Mycologie Médicale, Société Française de Physique, Association Française d’Histoire Économique, Société Française de Microbiologie. Liste mise à jour le 10 juillet 2019.
Introduction :
Suite à l’annonce le 1er février par le Premier Ministre d’une loi de programmation pluriannuelle de la recherche, un collectif de 23 sociétés savantes couvrant la majorité des grands champs disciplinaires a lancé une consultation sur les attentes de la communauté académique vis-à-vis de cette loi.
Sur la base des réponses à une première enquête générale, quatre enquêtes thématiques ont été mises en ligne entre le 19 avril et le 18 mai 2019 :
- Financement institutionnel de la recherche publique
- Emploi scientifique
- Organisation administrative de la recherche
- Relations entre recherche publique et société
Chaque sondage était ouvert pendant une période de 2 semaines ou plus. En tout, 9000 réponses ont été collectées grâce aux cinq enquêtes. L’analyse des adresses courriel laissées par 41% des répondant.e.s permet d’estimer qu’environ 5500 personnes ont participé à au moins une des enquêtes. La grande majorité des répondant.e.s étant chercheurs ou enseignant.e.s-chercheurs titulaires (>80%), on peut estimer qu’entre 5 et 10% de la communauté de cette catégorie de personnels a contribué. Les femmes représentent environ 35% des répondant.e.s.
Ces propositions, ainsi que le cadrage financier qui les accompagne, ont été présentées aux trois groupes de travail mandatés par le Premier Ministre.
Les synthèses brutes, sans interprétation, des réponses obtenues à chacun de ces questionnaires sont consultables sur le site portail des sociétés savantes académiques. Le présent document complète ces synthèses, sur lesquelles il se base pour formuler des recommandations prioritaires.
Organisation de la recherche :
Constat :
Le paysage de la recherche publique est complexe, constitué d’établissements d’enseignement supérieur et d’organismes de recherche dépendant de plusieurs ministères. Aucune structure politique identifiée n’assure actuellement le pilotage stratégique de l’ensemble de la recherche publique. Les organismes de recherche nationaux ont des contours thématiques parfois fortement chevauchants, un mécanisme d’alliance d’organismes étant censé coordonner les recherches au sein de chacun des grands champs disciplinaires. La majorité des laboratoires de recherche, contractualisés avec un grand organisme de recherche, est multi-tutelles (ce qui rend leur gestion complexe), mais il existe aussi de nombreuses Equipes d’Accueil, inquiètes de la fin d’une labellisation nationale qui vient d’être décidée à leur propos.
Ce qui ressort des enquêtes :
L’organisation générale de la recherche est considérée comme excessivement morcelée, sans que des pistes consensuelles de fusion d’organismes émergent. L’action des Alliances ne convainc pas les acteurs. Le MESRI doit être renforcé et ses missions étendues à la coordination de l’ensemble de la recherche publique civile. La complexification et la rigidification des règles administratives et comptables au cours des 10 dernières années constituent un obstacle majeur à l’activité et à la motivation du monde académique. Elles impactent le dynamisme des actrices et acteurs et limitent le nombre de dépôts de demandes de financements internationaux, au niveau européen notamment.
Recommandations prioritaires :
1) Redonner du temps de recherche aux chercheurs et enseignant.e.s-chercheurs.
a. Abaisser le service annuel d’enseignement statutaire des EC de 192h à 150h équivalent Travaux Dirigés, le service actuel étant très largement supérieur aux pratiques internationales. Alléger les services d’enseignement des EC nouvellement recrutés (au-delà des 32 heures de décharge actuelles) et recruter un nombre plus élevé de chercheurs et EC permanent.e.s pour assurer aux étudiants un taux d’encadrement supérieur tout en limitant le recours excessif aux vacataires.
b. Augmenter le nombre de congés sabbatiques réguliers, comme cela se pratique dans les autres pays, et les possibilités de délégation temporaire des EC vers les grands organismes.
c. Limiter le gaspillage de ressources et le temps de recherche perdu à écrire des projets finalement non financés en renforçant conjointement le financement sur dotation des laboratoires et le taux de succès des appels à projets (voir section financement).
d. Renforcer le soutien technique et administratif aux chercheurs par recrutement d’ITAs/BIATSS.
2) Rétablir une confiance mutuelle entre administrations et chercheurs.
a. Remplacer la politique actuelle de contrôle des dépenses a priori des laboratoires, chronophage et jugé inefficace, par un contrôle a posteriori.
b. Rapprocher les mondes de plus en plus divergents des administrations déléguées et centrales d’une part, des laboratoires de l’autre. Créer des incitations à la mobilité des agents administratifs entre ces deux mondes.
3) Faciliter la gestion multi-tutelles des unités de recherches et laboratoires.
a. Donner une plus grande liberté aux laboratoires pour dépenser leurs crédits : possibilité de report des crédits non dépensés d’une année sur l’autre, utilisation possible des dotations comme salaires et gratifications de stages. Séparer la dotation aux laboratoires pour infrastructure de celle pour frais de recherche.
b. Assouplir les règles des marchés publics et augmenter les possibilités de dérogation, pour les frais de mission notamment.
c. Homogénéiser les logiciels de gestion entre organismes, en prenant soin de mettre les utilisateurs au centre du processus de conception de ces logiciels.
Financement de la recherche :
Constat :
L’investissement public dans la recherche publique civile stagne à environ 0.8% du PIB depuis plusieurs décennies, soit un déficit annuel d’environ 4,5Md€ par rapport aux objectifs de la stratégie de Lisbonne (1% du PIB dédié à la recherche publique civile). Plusieurs pays européens ont pourtant atteint cet objectif et continuent de renforcer leur investissement dans la recherche publique (exemple : loi de programmation décennale allemande promettant un accroissement annuel de +3%), menaçant la recherche française d’un décrochage par rapport à des pays comme l’Allemagne, les pays scandinaves, les États-Unis ou la Chine. Structurellement, un glissement important a de plus eu lieu au cours des 15 dernières années, le financement des recherches des laboratoires (hors salaires permanents et infrastructures) passant d’un système de financement majoritaire par dotation d’Etat à un système de financement majoritaire par contrats sur Appels à Projets (AAP).
Ce qui ressort des enquêtes :
Le manque de financements, attribué à une mauvaise compréhension par le monde politique du rôle crucial joué par le monde académique dans la société et l’économie, impacte fortement et négativement les activités des laboratoires publics. Un rééquilibrage entre crédits sur dotation d’État et crédits sur AAP est demandé, ainsi que la création d’instruments de financements sur AAP plus diversifiés. L’Agence Nationale de la Recherche est fortement critiquée pour le faible taux de succès de ses AAP et pour l’opacité de son fonctionnement.
Recommandations prioritaires :
1) Accroître significativement le budget de la recherche publique civile.
a. Respecter l’engagement pris par la France au conseil Européen de Lisbonne (Mars 2000) d’investir 3% du PIB dans la recherche dont 1% dans la recherche publique civile.
b. Aligner le taux de succès actuel des AAP de l’ANR (15%) sur celui des AAP de la DFG allemande, soit 30%.
c. Augmenter les dotations de base des laboratoires (demande de 93% des répondant.e.s), pour couvrir environ 70% de leurs frais de recherche (hors salaires permanents et infrastructure), et les allouer dans leur majorité sur une base pluriannuelle en début de contrat quinquennal.
2) Améliorer le fonctionnement de l’ANR
a. Adapter les appels à projets aux besoins propres des disciplines, et créer un AAP spécifique pour des contrats postdoctoraux pluriannuels en France.
b. Rendre plus transparent le processus de prise de décision stratégique, de désignation des membres des comités de sélection, et de décision de financement. Consulter régulièrement la communauté sur ses attentes.
3) Soutenir le montage des dossiers européens et la gestion des contrats obtenus.
a. Mettre en place des structures publiques efficaces d’aide au montage des dossiers de financement, souvent lourds, des AAP européens.
b. Simplifier la gestion administrative des contrats européens, optimiser les logiciels de justification RH et financières.
Emploi scientifique :
Constat :
Il y a une baisse d’attractivité pour les métiers de la recherche qui s’exprime notamment par une diminution nette du nombre d’inscriptions en première année de doctorat au cours des dix dernières années. Cette baisse est corrélée à une faible reconnaissance du doctorat en-dehors du monde académique, comparée aux autres pays de l’OCDE, et à une politique de rémunérations très inférieures, à la fois aux standards internationaux et aux rémunérations et à la progression de carrière des agents de rang A+ dans les autres ministères.
Ce qui ressort des enquêtes :
L’insuffisance des débouchés du doctorat dans les fonctions publiques hors ESR et dans le monde économique, une spécificité française, est un facteur aussi important de la perte d’attractivité des carrières académiques que le faible nombre d’emplois permanents dans la recherche publique. Le nombre de recrutements sur emplois permanents de chercheurs, EC, et personnels de soutien doit être augmenté. L’insuffisance des moyens donnés aux organismes pour appliquer la Loi Sauvadet a conduit à une perversion de l’esprit de la loi et à des situations très difficiles dans certaines disciplines, la biologie notamment. Le niveau de rémunération inférieur des agents dépendants du MESRI par rapport aux corps de même niveau des autres ministères (en particulier corps A+) ne peut être justifié et symbolise la faible reconnaissance de la recherche et de l’enseignement supérieur publics. Enfin, les unités éprouvent des difficultés à recruter des informaticiens contractuels du fait du faible niveau de rémunération proposé, comparé au secteur privé.
Recommandations prioritaires :
1) Augmenter les recrutements permanents dans tous les corps de l’ESR.
a. Mettre en place un plan pluriannuel de recrutement tenant compte des prévisions démographiques (départs en retraite et autres types de sortie, effectifs d’étudiants à accueillir, etc.) et budgétisé indépendamment de l’augmentation naturelle de la masse salariale (GVT).
b. Augmenter les recrutements dans les corps chercheurs et EC pour rajeunir la pyramide des âges, assurer un meilleur taux d’encadrement étudiant et réduire la précarité (notamment les vacations) dans les carrières académiques. Simplifier la gestion des carrières en généralisant à tous les corps A+ une structuration en classe normale et classe exceptionnelle, comme c’est le cas pour les Maîtres de conférences et Chargés de Recherche actuellement.
c. Renforcer les postes de soutien, administratifs d’une part pour libérer du temps de recherche pour les chercheurs et EC, et techniques d’autre part pour appuyer la réalisation des projets.
d. La mise en place d’un système de recrutement conditionnel des chercheurs et ECs, rendue possible par le caractère pluriannuel de la loi de programmation, pourrait être testée à titre expérimental dans certains établissements, l’extension éventuelle du dispositif étant sujette à un bilan de l’expérimentation. Cette expérimentation doit satisfaire aux conditions suivantes : i) que l’âge de titularisation ne soit pas reculé par rapport à la situation actuelle, ii) que les conditions à remplir pour une titularisation soient explicites au moment du recrutement, iii) que le recrutement initial se fasse sur concours, la titularisation sur examen, un poste étant systématiquement créé, iv) que ce dispositif ne concerne qu’une fraction limitée des recrutements (~5%).
2) Améliorer l’attractivité et le déroulement des carrières académiques
a. Créer des contrats doctoraux en nombre suffisant dans le secteur des SHS/lettres/arts, les thèses réalisées sans financement devant devenir exceptionnelles.
b. Aligner les rémunérations des personnels de la recherche publique et de l’enseignement supérieur sur la moyenne des rémunérations pratiquées dans les corps de rang équivalent des autres ministères.
c. Développer et simplifier les possibilités de mobilité temporaire (vraies années sabbatiques, voir supra) et permanentes (mutations géographiques) des chercheurs et enseignant.e.s/chercheur.e.s.
d. Octroyer aux chercheurs et EC nouvellement recrutés un budget recherche initial leur permettant de lancer leurs projets.
e. Mieux reconnaître la prise de responsabilités collectives (décharges d’enseignement, primes de fonction significatives, octroi de budgets de recherche pour compenser le temps investi au service de la communauté).
Relations entre recherche publique et société :
Constat :
L’article L111-1 du code de la recherche modifié en juillet 2013 codifie les missions de la recherche, sans préciser les moyens associés, ni les modalités d’évaluation des différentes missions. Un équilibre satisfaisant entre la liberté des recherches académiques, basée sur la notion d’excellence académique au service de la production de savoirs, et la réponse aux attentes de la société, privilégiée par les décideurs politiques, est actuellement difficile à trouver. Il est parfois reproché aux laboratoires publics d’être trop coupés de la réalité, ce qui peut inciter les décideurs politiques à piloter la recherche publique vers des recherches plus appliquées et contribuer au faible niveau de recherche partenariale en France. La trop faible représentation des docteurs dans les hautes fonctions publiques (environ 2% contre plus de 20% en Allemagne, États-Unis, etc.) et dans les entreprises (moins de 15% de docteurs parmi les chercheurs privés) est un fort handicap à la prise en compte des méthodes et résultats de la recherche publique par les décideurs politiques et économiques. Enfin, l’empilement récent de nombreux dispositifs et structures publics de transfert de technologie aux missions insuffisamment définies, n’a pas eu l’effet désiré sur le développement de la recherche partenariale.
Ce qui ressort des enquêtes :
Les répondant.e.s à l’enquête sur les relations entre recherche publique et société sont dans leur grande majorité impliqués dans des actions de médiation scientifique auprès du public, de recherche partenariale avec la R&D privée, ou d’interactions avec le monde politique. L’investissement dans les missions de transmission et de transfert est néanmoins freiné par le fardeau administratif, auquel s’ajoute pour les EC un lourd service d’enseignement. Les acteurs de l’ESR considèrent que leurs missions principales ou importantes sont multiples, incluant la production de connaissances pour le bien commun, la formation et l’éducation des citoyens tout au long de leur vie, l’aide à la prise de décision politique, la formation des cadres de l’Etat et du monde académique, et enfin le transfert des découvertes académiques vers le monde économique. Il ne faut donc pas considérer l’interaction de la recherche publique avec la société sous le seul angle des relations avec le monde économique. L’extension récente des missions du Conseiller Scientifique en Chef du Canada constitue une expérience intéressante de meilleure intégration de la recherche publique dans la société et dans le processus de décision politique.
Recommandations prioritaires :
1) Mieux valoriser le doctorat dans la société pour améliorer la diffusion de la méthode et de la culture scientifiques dans la société et le transfert des résultats de la recherche publique vers le monde économique.
a. Profiter de la réforme en cours des hautes fonctions publiques (HFP) pour porter le pourcentage de recrutement de docteur.e.s dans les HFP à 20%, comme dans la majorité des pays de l’OCDE.
b. Conditionner l’octroi d’aides publiques (directes ou par crédit d’impôt) à la R&D privée à un taux de recrutement en CDI de jeunes docteurs.
c. Développer les contrats CIFRE pour atteindre 20% des thèses, en simplifier et accélérer la mise en place, en faire la promotion auprès des administrations, collectivités territoriales et associations éligibles.
d. Renforcer les moyens de suivi centralisés des carrières professionnelles des docteurs hors ESR (exemple, enquêtes IPdoc) pour les étendre au long terme (thèse + 10 ans). Favoriser l’intervention des docteurs hors ESR dans les écoles doctorales.
e. Conduire une réflexion sur la durée optimale de la thèse, en prenant en compte l’accroissement du temps passé en formations/missions complémentaires en plus du temps dédié à la recherche. Garantir aux doctorant.e.s le respect du droit du travail en matière de droit à congés, notamment en cas de maternité ou de congé parental.
2) Mieux reconnaître et prendre en compte dans la formation et les progressions de carrière l’implication des acteurs de la recherche publique dans des interactions avec la société.
a. Mieux définir les modalités de l’évaluation individuelle de l’ensemble des missions du monde académique, y compris la formation des citoyens, l’aide à la décision politique et la formation des cadres de l’Etat.
b. Généraliser la proposition par les établissements de missions doctorales complémentaires, rémunérées, de médiation scientifique.
3) Encourager les collaborations mutuellement bénéfiques entre recherche publique, administrations et collectivités locales, associations et R&D privée.
a. La recherche partenariale ne doit pas se limiter à la sous-traitance auprès des laboratoires ou plateformes technologiques publics. Elle doit inclure une vraie dimension collaborative dans le respect des missions de chaque partenaire.
b. Répartir justement la propriété intellectuelle entre partenaires publics et privés, simplifier et accélérer les procédures d’établissement de conventions de recherche.
c. Réfléchir aux moyens d’éviter la captation des meilleurs éléments par de grandes entreprises dans certaines disciplines (statistiques, big data, intelligence artificielle…), qui pourrait conduire à un assèchement de la recherche académique dans ces disciplines.
d. Mieux financer les laboratoires publics pour accroître leur attractivité auprès des partenaires économiques, ainsi que leur indépendance.
4) Simplifier le paysage et l’action des structures et des dispositifs de transfert technologique et d’incitation à la recherche partenariale.
a. Revoir la philosophie des dispositifs existants qui, à l’exception des contrats CIFRE, sont considérés peu efficaces et sont souvent méconnus des chercheurs.
b. Fusionner les structures locales ou nationales chevauchantes ou en compétition directe. A minima, créer un portail local unique d’accueil.
c. Editer un guide de bonnes pratiques pour le partage de propriété intellectuelle, accepté par tous, et dont le respect doit conditionner l’accès aux aides publiques.
d. Développer les réseaux informels qui relient les entreprises à la communauté scientifique, y compris pendant les études supérieures, pour que R&D privée, administrations, associations et recherche publique se connaissent et se comprennent mieux (séminaires, visite de laboratoires…).
e. Abandonner l’injonction de rentabilité faite aux structures de transfert technologique (exemple SATT).
Cadrage budgétaire :
Les propositions ci-dessus sont finançables dans le cadre des engagements internationaux de la stratégie de Lisbonne. Les postes budgétaires principaux sont :
Financement de la recherche (hors personnels permanents et infrastructures) :
Couvrir 70% des frais de recherche des laboratoires sur dotation de base : 600M€/an
Amener le taux de succès aux AAP de l’ANR à 30% : 600M€/an
Politique d’emploi :
Aligner les rémunérations des personnels de la recherche publique sur les corps équivalents des autres ministères : 2Md€/an
Recruter 2000 chercheurs, EC et ITA/BIATSS supplémentaires/an (sur 5 ans) : 300M€/an
Infrastructures :
Remise à niveau de l’immobilier universitaire et de la recherche publique :1Md€/an
Total : 4,5Md€/an
Il est à noter que ce chiffrage dépasse le cadre pur de la recherche publique, puisqu’une partie de la revalorisation salariale et des dépenses d’infrastructure concerne l’enseignement supérieur.
Concours Beautiful Science – Les lauréats
La Société Française de Physique et ses partenaires, dont la Société des Neurosciences, ont le plaisir de dévoiler les lauréat(e)s du concours Beautiful Science.
https://www.sfpnet.fr/oeuvres-laureates-du-concours-beautiful-science
Le thème était le suivant : “Montrer la science dans ce qu’elle a de plus beau et de plus élégant, de l’infiniment petit à l’infiniment grand !”. Plus de 360 propositions ont été reçues : photos, peintures, schémas, dessins, vidéos et sons provenant d’une grande diversité de profils. Pari réussi pour ce concours qui avait pour objectif de rassembler professionnels comme amateurs de science autours d’un thème fédérateur et enthousiasmant.
Activité cérébrale lors de tâches complexes révélée par la neuroimagerie ultrasonore ultrarapide

L’électrophysiologie, plus récemment les mesures optiques mais également les techniques d’imageries magnétiques, ont permis d’enregistrer des évènements associés directement ou indirectement à l’activité neuronale. Ces techniques bien que très performantes ont chacune des échantillonnages temporels et/ou spatiaux limités. C’est le cas spatialement de l’électrophysiologie qui par le nombre de micro-électrodes ainsi que la surface des contacts pour les enregistrements sont limités quant aux régions étudiées. Cette contrainte spatiale est également problématique pour les méthodes optiques du fait de la diffraction de la lumière qui limite la zone d’enregistrement de l’activité neuronale d’intérêt.
À l’opposé de ce spectre spatial, les techniques d’imageries fonctionnelles peuvent enregistrer une adaptation métabolique à l’échelle du cerveau dans son ensemble, mais avec des compromis de sensibilité et de résolution temporelle tout à fait limitante (de l’ordre de plusieurs secondes en comparaison de quelques millisecondes pour l’électrophysiologie).
Le développement récent de techniques d’imagerie ultrasonores neuro-fonctionnelles basées sur les mesures de doppler ultra-rapide (fUltrasound imaging) offrent un nouvel et unique moyen de quantifier les variations hémodynamiques cérébrales à haute résolution spatiale (100microns) et haute résolution temporelle (10msec). Chez le rongeur, cette technique a permis de révéler une sensibilité de signal sur bruit tout à fait intéressante. En revanche, la capacité de cette technique de révéler la dynamique spatiotemporelle de réseau dans le cadre de stimuli endogènes mis en jeu chez des animaux avec un cerveau de plus grande taille comme le primate lors de taches complexes n’a pas encore été démontrée.
Dans le cadre d’une étude récente nous avons démontré l’utilité de la technique d’imagerie ultrasonore capable de capturer instantanément (10ms) les changements métaboliques de région corticale et sous corticale chez le macaque effectuant des tâches cognitives complexes (Antisaccade, Estimation de durée etc…). De manière surprenante, la neuroimagerie ultrasonore est capable de suivre la propagation de l’information cérébrale d’une couche à une autre dans le cortex ainsi que d’apporter une information causale et directionnelle sur la propagation de l’activité cérébrale d’une zone à une autre.
Référence
Dizeux A, Gesnik M, Ahnine H, Blaize K, Arcizet F, Picaud S, Sahel JA, Deffieux T, Pouget P, Tanter M. Functional ultrasound imaging of the brain
reveals propagation of task-related brain activity in behaving primates. Nat Commun. 2019 Mar 28;10(1):1400. doi: 10.1038/s41467-019-09349-w.
Contact chercheurs
Pierre Pouget, INSERM 1127, CNRS 7225, Institut du Cerveau et de la Moelle épinière, Sorbonne Université, Paris, France
Mickael Tanter, Physics for Medicine, ESPCI, INSERM, CNRS, PSL Research University, Paris, France
Circuits thalamocorticaux de la prise de décision
La capacité à prendre une décision adaptée dans un environnement changeant fait intervenir de multiples régions cérébrales interconnectées. Le cortex préfrontal est l’un des sites primordiaux mais ces dernières années ont vu apparaître le rôle important des régions thalamiques. Actuellement, l’implication de ces circuits thalamocorticaux dans les fonctions cognitives est reconnue comme déterminante, particulièrement en ce qui concerne les liens entre le cortex préfrontal (PFC) et le thalamus médiodorsal (MD). Le cortex préfrontal se caractérise néanmoins par une importante hétérogénéité anatomique, à laquelle fait écho la diversité des projections thalamocorticales issues du MD : différentes populations neuronales thalamiques innervent les différentes régions préfrontales.
Récemment, l’équipe Décision et Adaptation de l’INCIA (Institut de Neurosciences Cognitives et Intégratives d’Aquitaine, UMR 5287, Bordeaux) a pu établir que les connexions entre le mPFC et le MD sont essentielles pour la capacité à prendre une décision adaptée en usant d’approches pharmacogénétiques avancées chez le rat. Ces résultats lèvent un voile partiel sur la contribution fonctionnelle des connections entre cortex préfrontal et thalamus en se focalisant spécifiquement sur le mur médian du cortex préfrontal. Néanmoins, le cortex orbitofrontal (OFC) fait également l’objet d’une innervation importante du MD et d’une autre région thalamique largement méconnue, le thalamus submédian.
Pour donner un éclairage plus complet sur le rôle fonctionnel de ces circuits, l’équipe a donc déconnecté l’OFC de l’une et l’autre de ses afférences thalamiques et évalué l’impact de ces manipulations sur la capacité à prendre une décision fondée sur la valeur courante du but chez le rat. Ceci est classiquement effectué en utilisant une procédure de dévaluation spécifique du but, après un apprentissage instrumental initial dans lequel les animaux apprennent que l’appui sur deux leviers différents permet d’obtenir une récompense alimentaire spécifique. Après cette phase, la procédure de dévaluation consiste à fournir à l’animal l’une de ces deux récompenses à volonté, et à procéder à un test de choix entre les deux leviers immédiatement après. Lors de ce test, les leviers sont inactifs et les choix des animaux sont donc fondés uniquement sur leur représentation de la valeur courante de la récompense : des animaux « normaux » répondent majoritairement sur le levier correspondant à la récompense non dévaluée. De façon intéressante, les déconnections entre cortex orbitofrontal et thalamus médiodorsal ou submédian n’ont pas produit d’effet, suggérant un rôle spécifique du circuit mPFC-MD (Alcaraz et al., 2018). Néanmoins, après une phase d’apprentissage instrumental supplémentaire dans lequel la flexibilité cognitive des animaux est sollicitée, en inversant les contingences entre levier et nourriture qui lui est associée, un résultat bien différent apparaît. Alors que la déconnection de l’OFC et du MD est toujours sans effet, déconnecter l’OFC de son autre afférence thalamique, le thalamus submédian, produit un déficit spécifique lors du choix, suggérant que ces animaux ne sont pas capables de s’adapter au changement de contingences.
Ce résultat fait écho à une démonstration similaire de l’équipe ayant préalablement établi un rôle du thalamus submédian dans la capacité à mettre à jour les contingences Pavloviennes (Alcaraz et al., 2015). De façon plus générale, pris dans leur ensemble, ces études soulignent que différents circuits thalamocorticaux semblent coopérer pour assurer le caractère flexible de l’action dirigée vers un but. Un enjeu important pour les années à venir sera de déterminer les mécanismes par lesquels les échanges fonctionnels entre ces circuits peuvent opérer.
Référence
A thalamocortical circuit for updating action-outcome associations (2019) Fresno V, Parkes SL, Faugère A, Coutureau E*, Wolff M*. *Contributed equally.
Elife. 2019 Apr 23;8. pii: e46187. doi: 10.7554/eLife.46187.
Pour aller plus loin
Thalamocortical and corticothalamic pathways differentially contribute to goal-directed behaviors in the rat (2018) Alcaraz F, Fresno V, Marchand AR, Kremer EJ, Coutureau E, Wolff M.
Elife. 2018 Feb 6;7. pii: e32517. doi: 10.7554/eLife.32517.
https://lejournal.cnrs.fr/nos-blogs/aux-frontieres-du-cerveau/comment-le-cerveau-decide
http://archives.cnrs.fr/presse/article/4227
Contact chercheurs
Equipe Décision et Adaptation, Institut de Neurosciences Cognitives et Intégratives d’Aquitaine (INCIA), UMR5287, Bordeaux
Effet paradoxal des antidéprésseurs pendant le développement : une cible inattendue dans le cortex préfrontal
Les inhibiteurs de la recapture de la sérotonine (IRS) sont les antidépresseurs les plus prescrits dans la prise en charge des états dépressifs et anxieux. Leur succès tient en particulier au fait qu’ils ont peu d’effets secondaires. Cependant il pourrait en être autrement pendant le développement. C’est tout au moins ce que démontrent les études précliniques chez le rongeur, révélant un effet paradoxal des IRS quand ils sont administrés pendant le développement. En effet, pendant une période critique du développement postnatal, les IRS favorisent l’émergence de symptomes anxieux et dépréssifs chez l’adulte. Les mécanismes de cet effet paradoxal sont mal connus.
Une publication récente de l’équipe du Dr Gaspar à l’institut du Fer à Moulin (Inserm, Sorbonne Université) apporte un éclairage nouveau sur cette question. Mariano Soiza-Reilly et collaborateurs viennent de montrer que les IRS perturbent le développement des circuits reliant le cortex préfrontal au raphe. Ce sont des circuits cruciaux dans la réponse au stress notamment pour modérer les réponses anxieuses. Soiza-Reilly et al. montrent que le développement de ces circuits préfrontaux se poursuit pendant les premières semaines de vie postnatale chez le rongeur, et qu’il est durablement modifié par les IRS, administrés pendant cette même période.
Les chercheurs ont d’abord fait l’observation inattendue d’une expression transitoire du transporteur de la sérotonine (SERT) dans une sous-population de neurones pyramidaux du cortex prefrontal de la souris. Cette observation prolonge des observations antérieures de l’équipe montrant que l’expression du SERT est plus large pendant le développement que chez l’adulte, s’étendant en particulier en dehors des neurones sértoninergiques du raphe. Dans la présente étude, le marquage génétique des neurones SERT+ préfrontaux a permis leur identification transcriptomique et anatomique précises. Il s’agit de neurones pyramidaux, glutamatergiques dont les projections sous-corticales ciblent le thalamus et différents noyaux du tronc cérébral, le raphe en particulier. Ils forment des synapses excitatrices sur les neurones sérotoninergiques et Gabaèrgiques du raphe. Utilisant des approches combinées de pharmacologie et de génétique, les chercheurs ont ensuite pu démontrer que l’invalidation de SERT spécifiquement dans le cortex pendant une période critique, était nécessaire et suffisante pour induire une exubérance de synapses excitatrices dans différentes cibles sous corticales (thalamus et raphe). Pour éclairer le lien de ces circuits avec le comportement anxieux et dépressif provoqué par l’administration développementale d’IRS les auteurs ont utilisé des approches pharmacogénétiques. Celles ci ont permis de montrer que les neurones SERT+ du cortex préfrontal modulent effectivement les réponses au stress dans des tests classiques d’anxiété et de dépréssion (test de conflit, nage forcée).
Au total, cette étude montre clairement que les IRS ont des cibles cellulaires différentes pendant le développement et chez l’adulte. L’expression transitoire de SERT dans une sous-population de neuones du CPF, permet le contrôle local des taux de 5HT et régule ainsi finement la synaptogénèse. Une des question posée par cette observation est de déterminer quels récepteurs 5-HT sont impliqués dans ces effets sur la synaptogénèse. Une autre question importante est de savoir si ces données sont transposables au développement humain. Certaines données suggèrent que c’est bien le cas. Ainsi, l’analyse transcriptome du cerveau de foetus humain (Allen Brain Atlas) indique que le SERT est bien exprimé de manière transitoire dans le cortex préfrontal chez l’homme et des observations morphologiques faites dans l’équipe montrent un marquage des axones corticaux frontaux dans des cerveaux d’embryons humains de 11 semaines. Il est donc probable que des phénomènes analogues existent chez l’homme mais une echelle temporelle différente du développement.
Références
Soiza-Reilly M., Meye FJ. ,Olusakin O , Telley L, Petit E, Chen X, Mameli M, Jabaudon D, Sze J-Y, Gaspar P. SSRIs target prefrontal-raphe circuits during development to modulate synaptic connectivity and emotional behavior ; Mol Psychiatry. 2019 Jan 10. doi: 10.1038/s41380-019-0349-9.
Contact chercheur
Institut du Fer à Moulin, Paris, France
Un point clé de la théorie de l’apprentissage démontré grâce à l’optogénétique

Le cerveau apprend de manière constante pour nous permettre d’améliorer nos actions en fonction de nos expériences. Plusieurs théories visent à rendre compte de cette propriété fondamentale, l’une des plus populaires étant l’apprentissage par renforcement, utilisée aussi en intelligence artificielle. Cette théorie postule que l’apprentissage émerge grâce à un renforcement spécifique des connections entre les neurones qui sont actifs durant un événement, une action ou une suite d’événements et d’actions menant à une récompense. Un des point clé de cette théorie est que plus les neurones impliqués sont actifs, plus le renforcement des connections est rapide et solide. Ainsi les événements qui activent le plus fortement notre cerveau devraient être appris de manière prioritaire par rapport à d’autres événements.
Afin de démontrer que ce principes guide effectivement l’apprentissage biologique, l’équipe de Brice Bathellier (Institut des Neurosciences Paris Saclay) a utilisé deux méthodes optiques permettant de suivre et de modifier l’activité de larges ensembles de neurones définis génétiquement (optogénétique). Ils ont pu ainsi montrer que la quantité d’activité générée dans le système auditif corrèle avec la vitesse d’apprentissage lorsque des souris apprennent à associer des sons avec une récompense. Dans un deuxième temps ils ont pu faire apprendre aux souris à associer, non plus des sons, mais des activations précises, artificielles du système auditif. Grâce à cette manipulation, ils ont pu montrer un lien causal direct entre la quantité d’activité générée et la force du renforcement. Ainsi les neurones les plus actifs lors d’un événement sont bien ceux sélectionnés en priorité par les mécanismes d’apprentissage.
Référence
Ceballo et al., Cortical recruitment determines learning dynamics and strategy. Nature Communications, 10: 1479 (2019)
https://rdcu.be/budts
Contact Chercheur
Brice Bathellier
Paris-Saclay Institute of Neuroscience (NeuroPSI)
Department for Integrative and Computational Neuroscience (ICN)
UMR9197 CNRS/University Paris Sud
CNRS, Bldg. 32/33
1 Av. de la Terrasse, 91190 Gif-sur-Yvette, France
www.bathellier-lab.org
phone: +33 1 69823408
mail : bathellier@unic.cnrs-gif.fr
A key point of learning theory demonstrated through optogenetics
The brain learns constantly to improve our actions according to our experiences. Several theories aim to account for this fundamental property, one of the most popular being reinforcement learning, also used in artificial intelligence. This theory postulates that learning emerges through a specific reinforcement of connections between neurons that are active during an event, an action, or a sequence of events and actions leading to a reward. One of the key points of this theory is that the more active involved neurons are, the faster and more solid the reinforcement of connection is. Thus the events that most strongly activate our brain should be learned in priority with respect to other events.
In order to demonstrate that this principle actually guides biological learning, the team of Brice Bathellier (Paris Saclay Institute of Neuroscience) used two optical methods to read and modify the activity of large sets of genetically defined neurons (optogenetics). They were able to show that the amount of activity generated in the auditory system correlates with the speed of learning when mice learn to associate sounds with a reward. In a second experiment, they were able to teach mice to associate, no longer sounds, but precise, artificial activations of the auditory system. Through this manipulation, they were able to show a direct causal link between the amount of activity generated and the strength of reinforcement. Thus, the most active neurons during an event are those selected in priority by the learning mechanisms.
Des vagues corticales pour façonner la représentation du mouvement visuel
Comment le cerveau relie-t-il les informations visuelles dans l’espace et le temps ? Les illusions visuelles fournissent un paradigme expérimental pour étudier ces processus. Lorsque deux points sont présentés de manière statique et séquentielle à des positions différentes, l’observateur perçoit le mouvement d’un seul point allant d’une position à l’autre : le mouvement apparent. Pour de grandes séparations spatio-temporelles, le système visuel est mis au défi de relier ces informations et ainsi garder la trace de l’identité de l’objet le long du chemin du mouvement apparent, également connu sous le nom de “problème de correspondance”. Des chercheurs du CNRS (Marseille & Gif-sur-Yvette) et d’Aix-Marseille Université ont utilisé l’imagerie optique des colorants sensibles au potentiel de membrane dans le cortex visuel primaire (V1) de singes éveillés, combinée à la modélisation computationnelle, pour montrer que les connexions excitatrices et inhibitrices reliant les neurones séparés sur de longues distances à l’intérieur de V1, peuvent résoudre ce problème en liant les informations des deux stimuli dans l’espace et le temps. Ainsi deux vagues de propagation façonnent la représentation du mouvement illusoire, l’une facilitant la réponse dans la direction du mouvement, et l’autre, se déplaçant dans le sens opposé supprime la représentation résiduelle du premier stimulus. Les scientifiques proposent que cette vague suppressive soit un mécanisme de bas niveau pour résoudre les problèmes de correspondance ambigus et qu’elle contribuerait ainsi à encoder précisément la trajectoire du mouvement apparent à la surface de V1. Ces résultats, publiés dans The Journal of Neuroscience, démontrent quel rôle computationnel les vagues de propagation d’activité corticales peuvent jouer dans la représentation dynamique d’information sensorielle.
Référence
Chemla S, Reynaud A, di Volo M, Zerlaut Y, Perrinet L, Destexhe A & Chavane F. (2019).
Suppressive traveling waves shape representations of illusory motion in primary visual cortex of awake primate. Journal of Neuroscience, 2792-18.
Contact Chercheurs
Sandrine Chemla
Frédéric Chavane
Institut de Neurosciences de la Timone (INT), UMR 7289 CNRS & Aix-Marseille Université, Marseille
Courir ou manger du chocolat, un choix dicté par les récepteurs cannabinoïdes.

Les pathologies qui résultent de notre mode de vie sédentaire ont pour principale cause une inactivité physique, cette dernière étant souvent associée à une prise excessive de nourriture riche en sucres et/ou en gras. A l’opposé, une activité physique excessive aux dépens de la prise de nourriture peut également s’avérer nocive, comme l’illustrent des cas d’anorexie nerveuse. Ces données rendent donc cruciale la recherche des processus neurobiologiques contrôlant les motivations respectives pour l’activité physique et la prise alimentaire. Fruit de la collaboration entre des chercheurs de l’Inserm et du CNRS, une étude publiée le 07 Mars 2019 dans la revue JCI Insight révèle que les récepteurs cannabinoïdes CB1 jouent un rôle primordial dans le choix entre courir et consommer une nourriture chocolatée.
Les auteurs de ce travail avaient précédemment rapporté que les récepteurs des cannabinoïdes CB1, présents sur plusieurs types de neurones, jouent un rôle clef dans les performances lors d’une activité physique chez la souris. Cette conclusion était basée sur les performances réalisées par des animaux ayant un accès libre à une roue d’activité, un modèle qui ne permettait pas de distinguer le mécanisme mis en jeu (motivation, plaisir…). La motivation pour une récompense ne pouvant être estimée que par la mesure des efforts que l’individu, Homme ou animal, est prêt à fournir pour accéder à cette récompense, les chercheurs ont élaboré un modèle dans lequel chaque accès à la roue était conditionné par un effort préalable. Cet effort préalable consiste en l’introduction répétée du museau dans un réceptacle, condition sine qua none pour débloquer la roue. Après une période d’apprentissage de la tâche au cours de laquelle l’effort demandé était constant, les souris ont été confrontées à un test dans lequel l’effort demandé pour accéder à la roue a été augmenté de manière progressive. Exposées à ce test, des souris dépourvues de récepteurs CB1 ont montré un déficit de 80 % dans l’effort maximal qu’elles étaient prêtes à fournir pour accéder à la roue, et ce sans diminution des performances lors de leurs accès à la roue. Ce résultat indique que les récepteurs CB1 jouent un rôle majeur dans le contrôle de la motivation pour l’activité physique. L’utilisation d’autres souris génétiquement modifiées a également permis aux chercheurs de démontrer que ces récepteurs CB1 contrôlant la motivation pour l’exercice sont localisés sur des neurones GABAergiques.
Les chercheurs ont ensuite examiné si les récepteurs CB1 dans les neurones GABAergiques contrôlent la motivation pour une autre récompense, de la nourriture chocolatée (au même titre que les humains, les souris en raffolent même si elles sont bien nourries). Alors que les récepteurs CB1 jouent également un rôle dans la motivation pour la nourriture, mais à un degré moindre que dans la motivation pour l’activité physique, les récepteurs CB1 localisés sur les neurones GABAergiques ne sont pas impliqués dans la motivation pour la prise de nourriture chocolatée.
Dans notre vie quotidienne, nous sommes confrontés à un choix permanent entre plusieurs récompenses. Cette évidence a poussé les chercheurs à développer un modèle dans lequel, après apprentissage, les souris avaient le choix, moyennant les efforts décrits ci-dessus, entre une activité physique et de la nourriture chocolatée. La motivation pour l’activité physique l’a emporté sur la prise de nourriture chocolatée, à l’exception des souris dépourvues de récepteur CB1 de manière globale ou uniquement dans les neurones GABAergiques qui, elles, ont montré une préférence pour la nourriture.
Au-delà de ces résultats indiquant que le récepteur cannabinoïde est primordial pour la motivation pour l’activité physique, cette étude ouvre des perspectives pour pouvoir étudier les mécanismes neurobiologiques responsables d’augmentations pathologiques de cette motivation. Une illustration est fournie par l’anorexie nerveuse qui associe souvent une diminution de la motivation pour se nourrir à une augmentation de la motivation pour l’activité physique.
Source
The motivation for exercise over palatable food is dictated by cannabinoid type-1 receptors.
Muguruza C, Redon B, Fois GR, Hurel I, Scocard A, Nguyen C, Stevens C, Soria-Gomez E, Varilh M, Cannich A, Daniault J, Busquets-Garcia A, Pelliccia T, Caillé S, Georges F, Marsicano G, Chaouloff F.
JCI Insight. 2019 Mar 7;4(5). pii: 126190. doi: 10.1172/jci.insight.126190.
Contact chercheur
Francis Chaouloff
NeuroCentre INSERM U1215
Equipe “Endocannabinoïdes & NeuroAdaptation”
33077 Bordeaux
05 57 57 37 55
francis.chaouloff@inserm.fr
Des neurones de schéma dans le cerveau

Comment le cerveau se représente-t-il l’espace ? Des chercheurs du CNRS de Lyon et Grenoble ont observé l’activité cérébrale de macaques alors que ces animaux naviguaient dans des environnements 3D virtuels à la recherche d’une récompense. Ils ont montré que certains neurones d’une structure essentielle à la mémoire, l’hippocampe, permettent de mémoriser les détails des environnements (mémoire épisodique) tandis que d’autres encodent la logique d’organisation de l’espace, lorsqu’elle se répète. Ces « cellules de schéma » encodent le rôle fonctionnel attribué à des repères visuels plutôt que leur apparence. Ainsi, l’hippocampe pourrait représenter à la fois le caractère particulier d’expériences uniques en même temps que l’information commune à ces épisodes, réalisant un encodage compact des données à mémoriser. Ces résultats, publiés dans Science, montrent qu’une forme de pensée abstraite existe chez le macaque rhésus, et ouvrent ainsi des possibilités d’exploiter ce modèle animal pour faire progresser la compréhension de pathologies cliniques.
L’hippocampe est une structure du lobe temporal du cortex cérébral qui joue un rôle essentiel dans la mémoire des souvenirs (mémoire épisodique) ainsi que dans l’orientation spatiale. Des lésions de cette partie du cerveau, comme dans la maladie d’Alzheimer, mènent à des pertes mnésiques et de profondes désorientations. La collaboration entre des chercheurs de l’Institut des Sciences Cognitives Marc Jeannerod (Lyon, CNRS/Université de Lyon) et du GIPSA-lab (Grenoble, CNRS/UGA) a permis de préciser le rôle des neurones de l’hippocampe dans la mémoire spatiale chez le singe. Les animaux étaient plongés dans un environnement 3D virtuel (labyrinthe en étoile) où ils devaient trouver une récompense invisible en se repérant grâce à des éléments distants (des amers, p. ex. l’arbre de l’illustration). Après des semaines d’entraînement dans un environnement devenu familier, les animaux ont été testés dans des environnements à la géométrie identique mais dont les amers changent chaque jour. Quelques essais et erreurs sont alors suffisants aux animaux pour se repérer, démontrant ainsi leur compréhension de la tâche et de la structure de l’environnement : les animaux ont formé un schéma mental de l’environnement-type. Si beaucoup de neurones de l’hippocampe semblent coder des aspects uniques à chaque environnement, comme l’identité des amers, d’autres se comportent comme des “cellules de schéma” dont l’activité, une fois rapportée à la position de la récompense, est similaire dans tous les environnements. Ainsi, l’hippocampe pourrait représenter à la fois le caractère particulier d’expériences uniques en même temps que l’information commune à ces épisodes, réalisant un codage compact des données à mémoriser. Ceci est encore plus apparent si, au lieu de caractériser l’activité des neurones par rapport à la position de l’animal dans l’espace physique, on la repère dans un espace des états de la tâche, une représentation plus abstraite de la progression de l’animal vers son but, à l’origine utilisée en automatique ou en robotique, et prenant en compte la position, l’orientation et l’historique de navigation de l’animal. Les neurones de schéma utilisent donc tous ces indices pour construire une représentation fonctionnelle de l’environnement. Ce modèle animal pourrait être utile dans la compréhension de certaines pathologies cliniques.
Référence
Baraduc P, Duhamel JR, Wirth S (2019). Schema cells in the macaque hippocampus. Science, 2019 Feb 8; 363(6427):635-639.
Contact chercheurs:
Pierre Baraduc, GIPSA-lab, CNRS/U.Grenoble-Alpes, 11 rue des Mathématiques, 38402 Saint Martin d’Hères. 04 76 82 71 50. pierre.baraduc@gipsa-lab.fr
Sylvia Wirth, ISCMJ, CNRS/U.Lyon, 67 Bd Pinel, 69675 Bron. 04 37 91 12 32. sylvia.wirth@isc.cnrs.fr
La preuve est faite : Les champs magnétiques des structures profondes du cerveau sont visibles depuis la surface !
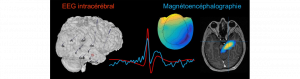
Une équipe pluridisciplinaire formée d’ingénieurs et de cliniciens (AMU, Inserm, AP-HM) vient de faire la démonstration qu’il est possible de détecter en surface des activités pathologiques se produisant dans des structures profondes du cerveau (Pizzo et al. Nat Comm 2018). Ces structures sont fortement impliquées dans des pathologies comme l’épilepsie ou certaines maladies neuro-dégénératives. Elles étaient considérées jusqu’à présent comme invisibles à partir de la surface, nécessitant l’implantation d’électrodes directement dans le cerveau par la technique d’EEG intracérébrale stéréotaxique (SEEG). Les chercheurs ont utilisé une combinaison unique d’enregistrements simultanés de magnétoencéphalographie (MEG) et de stéréo-électroencéphalographie (SEEG), et des méthodes avancées de traitement du signal. Ces résultats ouvrent de nouvelles possibilités dans l’étude non-invasive de la dynamique cérébrale, à la fois en clinique et en neurosciences fondamentales.
La MEG est une technique de pointe non-invasive utilisée pour cartographier les activités cérébrales, qui possède une excellente résolution à la fois spatiale et temporelle. La SEEG est une technique invasive utilisée lors du bilan préchirurgical des patients épileptiques, consistant à implanter des électrodes directement dans le cerveau. Très peu de centres au niveau mondial maîtrisent l’enregistrement simultané de ces deux méthodes, une prouesse technique qui a été rendue possible grâce à une collaboration rapprochée entre recherche et clinique. Ces enregistrements simultanés ont permis de confirmer la capacité de la MEG d’enregistrer le signal des zones du cerveau. Cela ouvre à terme la possibilité pour certains patients de se passer d’enregistrement invasifs, ce qui serait une grande avancée.
Les chercheurs et enseignants-chercheurs de l’Institut de Neurosciences des Systèmes (INS, Aix-Marseille Université et Inserm) et du service d’Epileptologie et de Rythmologie Cérébrale de l’assistance publique des hôpitaux de Marseille (AP-HM) ont ainsi pu montrer que des activités enregistrées avec des électrodes profondes dans l’hippocampe, l’amygdale et le thalamus produisent bien un reflet mesurable en surface sur les capteurs de MEG. Cela résout une controverse existante de longue date, car il est communément admis que des structures cérébrales aussi profondes et d’architecture complexe ne sont pas visibles directement, mais plutôt indirectement par propagation neuronale vers des structures plus superficielles. Grâce au traitement du signal, les chercheurs ont pu séparer les deux types d’activités, propagée et initiale, et ainsi démontrer que cette dernière est bien visible en surface.
Ces structures profondes du cerveau (en particulier du lobe temporal) sont impliquées à la fois dans le fonctionnement normal (mémoire, émotions) et dans le dysfonctionnement (épilepsie, maladies neurodégénératives) du cerveau. Cette découverte a donc des conséquences à la fois au niveau clinique, car elle suggère que l’on peut se passer d’implanter des électrodes pour diagnostiquer le cerveau, et au niveau des neurosciences, car elle ouvre la voie à de nouvelles études sur la dynamique spatio-temporelle des réseaux cérébraux.
Reference
Pizzo F, Roehri N, Medina Villalon S, Trébuchon A, Chen S, Lagarde S, Carron R, Gavaret M, Giusiano B, McGonigal A, Bartolomei F, Badier JM, Bénar CG. Deep brain activities can be detected with magnetoencephalography. Nat Commun. 2019 Feb 27;10(1):971. doi: 10.1038/s41467-019-08665-5.
Contact chercheur
Christian Bénar
Aix Marseille Univ, INSERM, INS, Inst Neurosci Syst, Marseille, 13005, France.
christian.benar@univ-amu.fr.