Tag: Actualité
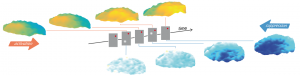
Des vagues corticales pour façonner la représentation du mouvement visuel
Comment le cerveau relie-t-il les informations visuelles dans l’espace et le temps ? Les illusions visuelles fournissent un paradigme expérimental…

Courir ou manger du chocolat, un choix dicté par les récepteurs cannabinoïdes.
Les pathologies qui résultent de notre mode de vie sédentaire ont pour principale cause une inactivité physique, cette dernière étant…

Des neurones de schéma dans le cerveau
Comment le cerveau se représente-t-il l’espace ? Des chercheurs du CNRS de Lyon et Grenoble ont observé l’activité cérébrale de macaques…
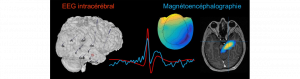
La preuve est faite : Les champs magnétiques des structures profondes du cerveau sont visibles depuis la surface !
Une équipe pluridisciplinaire formée d’ingénieurs et de cliniciens (AMU, Inserm, AP-HM) vient de faire la démonstration qu’il est possible de…

Les séquences neurales enchevêtrées sont indispensables à la formation de la mémoire !
De nombreuses fonctions cognitives semblent être sous-tendues au niveau cérébral par la formation de séquences d’activité neuronale, c’est-à-dire par l’activation…
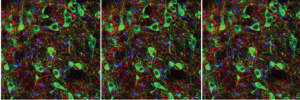
L’acétylcholine et les circuits neuronaux de la dépression
Selon l’Organisation Mondiale de la Santé, la dépression est un trouble mental courant affectant plus de 300 millions de personne…
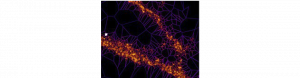
Changement nanoscopique, effet macroscopique : quand le récepteur NMDA régule l’adaptation synaptique
L’adaptation des synapses excitatrices est une des bases de la plasticité cérébrale, et implique le recrutement de récepteurs du glutamate…

Au delà des représentations sensorielles dans le cortex auditif primaire
Le cortex cérébral est classiquement décrit comme une chaine hiérarchique dans laquelle les aires primaires extraient et encodent les caractéristiques…
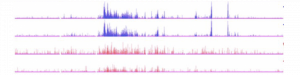
L’épigénétique au secours de la maladie d’Alzheimer !
La maladie d’Alzheimer (MA) est une maladie neurodégénérative affectant les fonctions de la mémoire et conduisant progressivement à une perte…
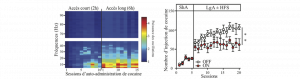
Soigner l’addiction à la cocaïne avec la chirurgie !
L’addiction est une maladie psychiatrique qui se caractérise par l’émergence de certains comportements pathologiques, tels que la perte progressive de…