Uncategorized
SNE Impact 2019
Chaque année, la Société de Neuroendocrinologie sélectionne une dizaine de publications majeures qui reflètent le dynamisme des différents domaines de recherche dans les laboratoires français de Neuroendocrinologie.
Le SNE Impact2019 présente la sélection des articles publiés en 2019.

Formation des neurones sensoriels : un processus dynamique finement régulé
Notre capacité à détecter et intégrer diverses modalités sensorielles telles que le toucher (mécanoception), l’état d’étirement de nos muscles et la position de notre corps dans l’espace (proprioception), la température (thermoception) ou la douleur (nociception) repose sur le système nerveux somatosensoriel. Dans le tronc, les récepteurs primaires de ce système, localisés dans les ganglions rachidiens dorsaux (GRD), forment une population neuronale hétérogène dont la diversité fonctionnelle est établie tôt au cours de l’embryogenèse. Les neurones des GRD se forment à partir de cellules souches (ou progéniteurs) issues de la crête neurale qui contribuent également à ce niveau aux neurones sympathiques, aux cellules gliales périphériques ainsi qu’aux mélanocytes (les cellules pigmentaires de la peau). Le choix entre ces différents destins cellulaires implique des modifications du potentiel développemental des cellules de la crête neurale en fonction du temps et/ou de l’environnement, notamment reflétées par l’induction de déterminants spécifiques à chaque lignage. Dans les progéniteurs somatosensoriels, les facteurs de transcription Neurog1 et Neurog2 ont été identifiés depuis longtemps comme acteurs essentiels à la différentiation des différents sous-types neuronaux en agissant de manière a priori indépendante et complémentaire. Il est en effet généralement admis qu’un premier groupe de progéniteurs se différencie sous le contrôle de Neurog2 pour générer d’abord les neurones mécanoceptifs et proprioceptifs, puis qu’un second groupe se différencie plus tard sous l’action de Neurog1 pour produire les neurones thermo/nociceptifs. Ce modèle est supporté par l’absence totale de GRD chez les animaux doubles-mutants Neurog1-/- ;Neurog2-/-, ainsi que par l’agénésie spécifique des neurones « tardifs » thermo/nociceptifs chez les simples-mutants Neurog1-/-. Cependant, il n’existe pas d’évidence directe conférant un rôle spécifique à Neurog2 dans la formation des neurones « précoces » mécano/proprioceptifs, aucune perte neuronale n’ayant été décrite chez les simples-mutants Neurog2-/-. De fait, la contribution individuelle de Neurog2 dans ce système restait étonnamment vague. Au cours d’une étude récente, nous avons mis en évidence que les GRD des mutants Neurog2-/- contiennent en fait un nombre globalement réduit de neurones de tous types, concernant non seulement la population mécano/proprioceptive mais aussi, de manière plus inattendue, la population thermo/nociceptive. Nous avons établi que ce phénotype général -bien que partiel- est très dynamique et est la conséquence de multiples défauts affectant l’intégrité de différents pools de progéniteurs qui surviennent tous pendant une courte fenêtre de temps au cours de laquelle Neurog2 régule transitoirement l’expression de Neurog1 et l’initiation des différentes vagues de différentiation. Nous avons notamment montré que durant cette période, certains progéniteurs dédiés au système somatosensoriel changent d’identité et adoptent un destin de mélanocytes, tandis que d’autres meurent par apoptose. Ainsi, lorsque Neurog1 est finalement induit chez les mutants Neurog2-/-, la neurogenèse est initiée de manière retardée, à partir d’un réservoir restreint de progéniteurs, expliquant à terme le déficit neuronal observé. Ces résultats ont ainsi révélé l’existence d’une période critique de vulnérabilité et de plasticité développementale parmi les populations de progeniteurs somatosensoriels. Ils ont aussi permis de déterminer qu’au cours de la formation des GRD, le rôle de Neurog2 est plus complexe et plus large qu’initialement envisagé.
Reference:
Neurog2 Deficiency Uncovers a Critical Period of Cell Fate Plasticity and Vulnerability among Neural-Crest-Derived Somatosensory Progenitors. Ventéo S, Desiderio S, Cabochette P, Deslys A, Carroll P, Pattyn A. Cell Rep. 2019 Dec 3;29(10):2953-2960
Contact chercheur:
Institute for Neurosciences of Montpellier, University of Montpellier, INSERM U1051, Montpellier, France.
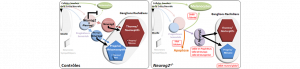
Syndrome de Rett : Lorsque la huntingtine sauve les neurones déficients
Une étude récente menée entre autres à Aix-Marseille Université et l’Inserm laisse entrevoir un espoir thérapeutique dans le syndrome de Rett ! En effet, les équipes de Jean-Christophe Roux (équipe de Neurogénétique Humaine) du Centre de Génétique Médicale de Marseille* et Frédéric Saudou du Grenoble Institut Neurosciences** ont élaboré une stratégie originale en axant leurs travaux sur la huntingtine, protéine impliquée dans la maladie de Huntington.
Le syndrome de Rett est un trouble neurologique grave qui affecte uniquement des filles avec une incidence de 1 sur 15 000 naissances. Ce n’est qu’entre 6 à 18 mois que les premiers signes apparaissent. Pour l’instant, il n’existe aucun traitement pour cette pathologie qui conduit à un polyhandicap sévère se traduisant par des troubles cognitifs, moteurs et autonomes. L’activation de la protéine Huntingtine, qui lorsque mutée est responsable de la maladie de Huntington (MH), améliore la physiopathologie et les symptômes dans des souris modèles du syndrome de Rett. En effet, l’élévation de son taux de phosphorylation permet de restaurer le transport endogène de BDNF – facteur neurotrophique dérivé du cerveau – essentiel au développement et au bon fonctionnement des neurones. De nombreuses études ont ainsi établi un lien direct entre la réduction des connexions neuronales et l’altération du taux de sécrétion du BDNF. Chez les malades atteints du syndrome de Rett, le gène responsable a été identifié en 1999. Il s’agit de MECP2 qui régule l’expression de milliers de gènes neuronaux dont celle du BDNF. Lorsque MCP2 est muté, l’expression du BDNF est diminuée de moitié, provoquant de nombreux problèmes de connectivité neuronale. En utilisant des approches génétiques et pharmacologiques, Jean-Christophe Roux et Frédéric Saudou ont montré que la phosphorylation de la huntingtine permet de pallier ce problème en augmentant le transport de BDNF dans des neurones modèles de Rett et améliore ainsi le fonctionnement des synapses. Des travaux antérieurs avaient identifié la molécule FK506, déjà utilisée en clinique pour éviter le rejet de greffe, comme augmentant la phosphorylation de la huntingtine. Les chercheurs ont alors montré que cette molécule était capable de rétablir le transport de BDNF dans les puces microfluidiques reproduisant les connexions dans les cerveaux Rett et améliorait la physiopathologie et les symptômes chez les souris modèle du syndrome de Rett.
Ces travaux, publiés dans le journal Embo Molecular Medicine le 08/01/2020, pourraient constituer une approche prometteuse pour le traitement des patientes atteintes du syndrome de Rett.
Référence : Ehinger Y, Bruyère J, Panayotis N, Abada YS, Borloz E, Matagne V, Scaramuzzino C, Vitet H, Delatour B, Saidi L, Villard L, Saudou F, Roux JC. Huntingtin phosphorylation governs BDNF homeostasis and improves the phenotype of Mecp2 knockout mice. EMBO Mol Med. 2020 Jan 8:e10889. doi: 10.15252/emmm.201910889.
Contact chercheur :
Jean-Christophe Roux, Équipe de NeurogénétiqueHumaine, MMG – Centre de génétique médicale de Marseille, Aix-Marseille Université – Inserm – UMR 1251, Faculté de Médecine de La Timone, Marseille.
Frédéric Saudou, Directeur du Grenoble institut des Neurosciences (GIN), Équipe “Dynamiques intracellulaires et neurodégénérescence”, Inserm, U1216, CHU Grenoble Alpes, Grenoble
Un nouvel acteur impliqué dans les troubles de la mémoire et de l’anxiété
Dans de nombreuses pathologies et maladies neurodégénératives, une augmentation d’une protéine appelée P2X4 est observée à la surface de cellules mais son rôle restait méconnu. L le groupe d’Eric Boué-Grabot, à l’IMN de Bordeaux a créé une souris qui permet d’augmenter le nombre de P2X4 à la surface de cellules afin de mimer la situation pathologique et en collaboration avec plusieurs équipes les chercheurs révèlent dans un travail publié dans Molecular Psychiatry, que le nombre accru de cette protéine dans les neurones crée une activité anormale dans le cerveau provoquant des troubles de la mémoire et une diminution de l’anxiété.
La “molécule de l’énergie” des cellules, l’ATP, est aussi libérée à l’extérieur des cellules et sert de messager entre les cellules. La protéine P2X4, récepteur de l’ATP, est exprimée dans de nombreuses cellules à travers tout l’organisme et plus particulièrement dans les neurones et les cellules gliales du cerveau. Cette protéine faiblement présente à la surface des cellules en conditions normales se révèle plus abondante chez certains neurones et/ou cellules gliales dans les maladies neurodégénératives telles que les maladies d’Alzheimer, de Charcot ou la sclérose en plaque mais aussi dans les douleurs chroniques et d’autres pathologies comme celles liées à la consommation d’alcool. Cette augmentation est aussi observée dans d’autres cellules de l’organisme en particulier dans des conditions inflammatoires comme l’asthme ou l’arthrose rhumatoïde suggérant que le récepteur P2X4 pourrait être un acteur-clé de nombreuses pathologies et par conséquent une cible thérapeutique potentielle.
Afin d’appréhender les rôles du récepteur P2X4, le groupe d’Eric Boué-Grabot, à l’IMN de Bordeaux a développé avec l’aide de la Clinique de la souris de Strasbourg a développé une lignée de souris permettant d’augmenter spécifiquement le nombre des récepteurs P2X4 à la surface de certaines cellules. Les résultats publiés dans le journal Molecular Psychiatry qui résultent d’une collaboration avec plusieurs équipes françaises CNRS et Inserm et des chercheurs de l’institut d’immunologie de Hambourg et du Neuro de l’université McGill de Montréal montrent que l’augmentation du nombre de récepteurs P2X4 à la surface des neurones dans une structure du cerveau (l’hippocampe) impliquée dans la mémoire et l’apprentissage provoque des déficits mnésiques ainsi qu’une diminution de l’anxiété des souris. Ils démontrent aussi que la présence accrue de ces récepteurs altère les processus cellulaires à la base de la mémoire. Ces travaux suggèrent que l’augmentation des récepteurs P2X4 à la surface de neurones observée dans la maladie d’Alzheimer pourrait contribuer aux déficits mnésiques et représenter ainsi une piste thérapeutique potentielle dans les maladies neuropsychiatriques.
Cette étude révèle aussi tout le potentiel de ce nouveau modèle murin car la modification génétique du récepteur P2X4 permet de visualiser directement l’augmentation du nombre de récepteurs P2X4 en situation pathologique. Ces souris devraient permettre dans le futur d’élucider le rôle de ces récepteurs dans différents modèles de pathologies non seulement du cerveau mais aussi du poumon, du cœur, ou lors d’infection et d’inflammation, situations pour lesquelles le nombre de ce récepteur augmente dans des cellules spécialisées.
Pour en savoir plus:
Increased surface P2X4 receptor regulates anxiety and memory in P2X4 internalization-defective knock-in mice.
Bertin E, Deluc T, Pilch KS, Martinez A, Pougnet JT, Doudnikoff E, Allain AE, Bergmann P, Russeau M, Toulmé E, Bezard E, Koch-Nolte F, Séguéla P, Lévi S, Bontempi B, Georges F, Bertrand SS, Nicole O, Boué-Grabot E.
Mol Psychiatry. 2020 Jan 8. doi: 10.1038/s41380-019-0641-8. [Epub ahead of print]
Contact chercheur: Eric Boué-Grabot ; Directeur de recherche CNRS
Institut des Maladies Neurodégénératives (IMN)(CNRS/Université de Bordeaux), Centre Broca Nouvelle Aquitaine
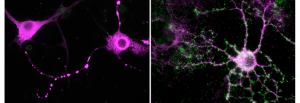
Comment la mémoire sociale émerge à l’adolescence ?
L’adolescence est reconnue comme une période critique du développement du cerveau avec des changements majeurs des capacités cognitives et un remodelage structurel important. Les mécanismes qui sous-tendent l’émergence de nouvelles fonctions cognitives sont cependant peu connus.
Dans de nombreuses structures cérébrales, la plasticité synaptique est plus forte pendant le développement postnatal, et cette plasticité joue un rôle important dans la mise en place des réseaux neuronaux. De façon surprenante, l’équipe de Rebecca Piskorowski et Vivien Chevaleyre (Institut de Psychiatrie et Neuroscience de Paris), en collaboration avec Laure Verret et Christophe Rey de l’équipe de Claire Rampon (Centre de Recherche sur la Cognition Animale, Toulouse), a mis en évidence une forme de plasticité synaptique qui n’est pas présente au début du développement postnatal chez la souris, mais qui apparaît à la fin de l’adolescence. Cette plasticité se produit sur les interneurones exprimant la Parvalbumine de la région CA2 de l’hippocampe, une région clé pour la formation de mémoire sociale. Les auteurs ont pu montrer que l’apparition de la plasticité est liée à la maturation du réseau périneuronal (PNN : PeriNeuronal Net), une différenciation de la matrice extracellulaire. Dans de nombreuses structures, l’augmentation du PNN au cours du développement contribue à la diminution de la plasticité synaptique et à la stabilisation des réseaux. Dans CA2, ce PNN à un rôle particulier en permettant l’apparition d’une plasticité synaptique. Ceci s’explique par le fait que le PNN permet de maintenir une signalisation trans-synaptique par l’intermédiaire de la Neuréguline 1 (exprimée par les neurones pyramidaux) et son récepteur ErbB4 (exprimé par les terminaisons axonales de interneurones Parvalbumine).
L’induction de cette plasticité par les récepteurs Delta des opioïdes, aussi exprimés au niveau des terminaisons des interneurones Parvalbumine, diminue la libération de GABA. Cette dépression à long-terme de l’inhibition (iLTD), change la balance en faveur de l’excitation et permet aux afférences de CA3 de recruter les neurones pyramidaux de CA2, ce qui n’est pas le cas lorsque l’inhibition est intacte. Comme l’activité des neurones pyramidaux de CA2 est nécessaire pour la formation de mémoire sociale, nous avons émis l’hypothèse que la iLTD pourrait avoir un rôle dans cette forme de mémoire. En accord avec cette idée, nous avons montré que la mémoire sociale apparaît au cours du développement en même temps que la plasticité synaptique sur les interneurones Parvalbumine. Par ailleurs, une dégradation du PNN dans CA2, ou l’utilisation d’un système viral pour diminuer l’expression des récepteurs Delta opioïdes dans CA2, ralenti la formation de la mémoire sociale.
Ces résultats procurent un lien entre l’apparition d’une plasticité synaptique à la fin de l’adolescence et l’émergence d’une nouvelle fonction cognitive.
Référence : Domínguez S, Rey CC, Therreau L, Fanton A, Massotte D, Verret L, Piskorowski RA, Chevaleyre V. Maturation of PNN and ErbB4 Signaling in Area CA2 during Adolescence Underlies the Emergence of PV Interneuron Plasticity and Social Memory. Cell Reports. 2019 Oct 29;29(5):1099-1112.e4. doi: 10.1016/j.celrep.2019.09.044.
Contact chercheur :
Rebecca Piskorowski/Vivien Chevaleyre : Institut de Psychiatrie et Neuroscience de Paris, INSERM U1266, Paris
Laure Verret : Centre de Recherche sur la Cognition Animale, Université Paul Sabatier, Toulouse
L’origine commune des cellules souches et des cellules épendymaires multiciliées

De nouveaux neurones sont générés tout au long de la vie dans le cerveau des mammifères. Les cellules souches à l’origine de ces neurones sont localisées dans des niches neurogéniques à proximité des ventricules latéraux. La compréhension des mécanismes qui régulent la formation de ces cellules est d’un grand intérêt puisqu’elle permettra à la fois d’élucider l’origine de certaines tumeurs, mais aussi d’élaborer des stratégies régénératives à partir de ces cellules souches.
La niche neurogénique adulte est composée de deux types de cellules gliales : les cellules souches astrocytaires qui conservent une capacité proliférative, et les cellules épendymaires multiciliées qui forment une barrière épithéliale entre les cavités ventriculaires et le parenchyme. Ces cellules épendymaires sont post-mitotiques et dotées de nombreux cils motiles, dont les battements coordonnés permettent la diffusion des morphogènes et des facteurs de croissance, et l’élimination des toxines contenus dans le liquide céphalo-rachidien (LCR) et à la surface des parois ventriculaires. Mis à part leur localisation au sein de la niche neurogénique, tout oppose donc ces deux types cellulaires puisqu’elles ne partagent ni leur morphologie, ni leur fonction. De façon surprenante, notre publication récente a permis de révéler qu’elles ont une chose en commun : leur mère !
Par des techniques de traçage génétique à long terme (plusieurs semaines), nous avons suivi des progéniteurs embryonnaires et etudié leurs descendances. La technique Brainbow colore de façon unique un grand nombre de clones qui peuvent être distinguables par l’analyse automatique informatisée des couleurs des cellules de la niche neurogénique. A l’inverse, la technique MADM (Mosaic Analysis of Double Markers) confère à un petit nombre de progéniteurs, la capacité de transmettre les couleurs verte et rouge à chacun de ses lignages fils. Le faible nombre de cellules marquées par la technique MADM permet de suivre sans ambiguité les modes de division (symmétrique ou asymmétrique), le nombre de division, la distance entre les cellules d’un clone, et l’identité des cellules. L’analyse des nombreux clones obtenus grâce à la technique Brainbow permet quant à elle d’obtenir des résultats statistiques robustes. Nos observations montrent que les cellules de la niche neurogénique adulte sont issues de la division asymmétrique d’un progéniteur embryonnaire qui produirait d’abord les cellules souches maintenues à l’état quiescent, puis les cellules épendymaires post-mitotiques. Nous avons de plus montré le rôle déterminant des protéines de la famille Geminin sur la composition des clones et donc sur le destin des progéniteurs embryonnaires. La famille Geminin est composée de 3 membres, connus pour réguler la réplication de l’ADN au cours du cycle cellulaire. GemC1 et MCIDAS, qui participent au complexe de pré-réplication de l’ADN, favorisent la production des cellules épendymaires au détriment des cellules souches adultes. A l’inverse, Geminin, qui inhibe la re-réplication de l’ADN en phase G2 du cycle cellulaire favorise la production des cellules souches adulte par division symmétrique des progéniteurs embryonnaires. Les cascades d’évenements par lesquels les protéines Geminin orientent le destin des progéniteurs sont inconnues et il reste à vérifier si ces facteurs agissent via la réplication de l’ADN et le cycle cellulaire ou de façon indépendante. A terme, ces recherches permettront d’élucider l’origine des cellules souches neurales adultes et de certaines tumeurs cérébrales.
Référence :
Ortiz-Álvarez G, Daclin M, Shihavuddin A, Lansade P, Fortoul A, Faucourt M, Clavreul S, Lalioti ME, Taraviras S, Hippenmeyer S, Livet J, Meunier A, Genovesio A, Spassky N. Adult Neural Stem Cells and Multiciliated Ependymal Cells Share a Common Lineage Regulated by the Geminin Family Members. Neuron, 102(1):159-172.
Contact :
Institut de Biologie de l’Ecole Normale Supérieure (IBENS), Ecole Normale Supérieure, CNRS, INSERM, PSL Université Paris, 75005 Paris, France.
Un neurone pour une mémoire
Le phénomène de neurogenèse adulte présent chez un certain nombre de mammifères fournit un apport permanent de nouveaux neurones à certaines régions du cerveau et plus particulièrement au système olfactif. Nous savions que ces nouveaux neurones étaient impliqués dans la formation et le maintien de la mémoire olfactive. Il restait cependant à déterminer comment ce renouvellement continu de nouveaux neurones pouvait d’une part assurer la stabilité de la mémoire déjà acquise et d’autre part préserver une flexibilité pour la formation de nouvelles mémoires.
Dans cette étude, nous révélons que lorsque le délai entre deux apprentissages olfactifs est court, les nouveaux neurones qui permettent le premier apprentissage sont très sensibles aux interférences et de ce fait, sont appelés à mourir lors du deuxième apprentissage, ce qui conduit à l’oubli de cette première mémoire. Dans ce contexte, nous avons utilisé la technique d’optogénétique qui permet grâce à la lumière de moduler sélectivement l’activité des neurones ciblées et nous montrons que si l’activité de ces nouveaux neurones est bloquée, la mémoire du premier apprentissage est affectée sans avoir de conséquence sur celle formée lors du deuxième. Ce résultat suggère que ces nouveaux neurones ne pourraient servir de support qu’à un seul souvenir olfactif. Par contre, lorsque l’information mémorisée reste utile à l’animal, au-delà de phase d’apprentissage, les nouveaux neurones survivent et la mémoire est conservée. Enfin, lorsqu’un délai plus long est permis entre les deux apprentissages, une résilience accrue des nouveaux neurones aux interférences est observée et les mémoires acquises successivement sont conservées. En conclusion, les nouveaux neurones réguleraient le caractère transitoire ou persistant d’une mémoire en fonction du délai entre deux apprentissages et de la pertinence des informations apprises.
Référence:
Jérémy Forest, Mélissa Moreno,Matthias Cavelius, Laura Chalençon, Anne Ziessel, Joëlle Sacquet, Marion Richard, Anne Didier and Nathalie Mandairon. Short-term availability of adult-born neurons for memory encoding. Nat Commun. 2019; 10: 5609. Dec 6. doi: 10.1038/s41467-019-13521-7.
Contact chercheur:
INSERM, U1028; CNRS, UMR5292; Lyon Neuroscience Research Center, Neuroplasticity and Neuropathology of Olfactory Perception TeamClaude Bernard University-Lyon1 and University of Lyon, Lyon, F-69000, France.
Prix Inserm 2019 – Hervé Chneiweiss, Prix OPECST-Inserm pour l’impact sociétal

Avec ce prix, l’Institut récompense les efforts de valorisation de la recherche et sa capacité à être véritablement en dialogue avec les attentes de la société et les questions des citoyens sur leur santé. Président du comité d’éthique de l’Inserm, Hervé Chneiweiss est reconnu pour son travail sur les astrocytes dont il a démontré les fonctions. Ses travaux, à la frontière de la neurologie et de la génétique l’ont très vite amené à s’intéresser à la bioéthique et à la question de la place de la science dans la société. Conseiller au cabinet du ministre de la Recherche de 2000 à 2002, il a notamment été en charge de la première révision des lois de bioéthique. L’Inserm, ainsi que l’’Office parlementaire d’évaluation des choix scientifiques et techniques, dont il a été membre du comité scientifique, souhaitent rendre hommage à la carrière de celui qui, élu en juillet à la présidence du comité international de bioéthique de l’Unesco, porte désormais cet engagement hors de nos frontières.
Urgence Horizon Europe : où sont les neurosciences ?

Bien que le mot « Cerveau » soit souligné à quelques endroits, la thématique « Brain Health » et la recherche sur le cerveau ne sont pas mentionnées dans le texte de partenariat de la Commission européenne pour la prochaine période de programmation intitulé Horizon Europe.
Pour la dernière fois probablement, le texte d’orientation vers un éventuel plan stratégique Horizon Europe est ouvert pour consultation jusqu’au 17 novembre sur le site de l’Union européenne.
À ce stade, les partenariats sont répertoriés pour le cluster Santé et il est entendu que les partenariats non répertoriés ne seront pas mis en œuvre dans la première moitié d’Horizon Europe.
C’est pourquoi nous vous demandons de vous exprimer pour la recherche sur le cerveau, et nous suggérerons de déposer des remarques sur l’absence d’appels à projets sur les recherches collaboratives sur le cerveau et le manque de structuration dans le domaine de la recherche en neurosciences dans le texte d’Horizon Europe.
Nous attendons avec impatience toute action visant à sauver Brain Health et la recherche en neurosciences.
Merci de répondre dans ce sens au questionnaire.
L’isolation des calculs corticaux pendant les ondes delta permet la consolidation de la mémoire
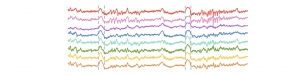
Pendant la majeure partie de notre temps de sommeil, notre cortex alterne entre des périodes
d’intense activité et des périodes de silence généralisé. Ces périodes de silence forment de grandes
déviations sur les enregistrements électro-encéphalographiques, qu’on nomme « ondes delta », et qui
jouent un rôle fondamental dans la mémoire : elles permettent de stabiliser les souvenirs à long
terme, notamment grâce à leur couplage avec d’autres rythmes cérébraux. Pourtant, les mécanismes
sous-jacents demeurent énigmatiques : comment des périodes de silence généralisé permettent-elles
aux circuits corticaux de se réorganiser pour consolider la mémoire ?
Pour bien prendre la mesure de cette énigme, nous devons revenir un instant sur ce que nous savons
de l’activité du cerveau pendant le sommeil à ondes lentes. La plupart de nos connaissances sur les
mécanismes fins nous viennent de travaux menés chez le rongeur. Nous savons tout d’abord que
l’hippocampe, la structure cérébrale qui permet entre autres de former de nouveaux souvenirs, se
réactive spontanément en générant une activité semblable à celle de l’éveil : comme si l’animal
« rêvait » de ce qu’il vient de vivre pendant l’expérience scientifique à laquelle il a participé. Ces
informations sont transmises au cortex, qui répond en activant des sous-groupes de neurones bien
spécifiques. Souvent, ce dialogue est suivi d’une onde delta et d’une activité rythmique appelée
« fuseau de sommeil » : c’est à ce moment semble-t-il que les circuits corticaux se réorganisent pour
former des souvenirs stables. Mais alors, pourquoi le dialogue hippocampo-cortical est-il interrompu
par une période de silence (onde delta) juste avant que les circuits corticaux ne puissent prendre en
compte ces échanges pour se réorganiser ? Ce silence n’efface-t-il pas les informations pertinentes ?
En examinant les ondes delta de plus près, nous avons tout d’abord constaté que contrairement à
l’idée généralement acceptée par la communauté scientifique, le cortex ne devenait pas totalement
silencieux : à chaque onde delta, un petit nombre de neurones sans cesse changeant restaient actifs.
Et il ne s’agissait pas d’un simple bruit de fond : de façon surprenante, nous avons découvert que
ces neurones formaient ce que les chercheurs nomment des « assemblées » — des ensembles de
neurones qui s’activent ensemble et de manière répétée pour permettre au cortex de coder des
informations. Plus étonnant encore, ces assemblées se formaient en réponse aux réactivations
spontanées de l’hippocampe. Cette observation inattendue suggérait qu’elles pouvaient être
impliquées dans la consolidation de la mémoire. Et de fait, ces assemblées contenaient
principalement des neurones qui avaient été particulièrement impliqués dans la tâche de
mémorisation que les rats avaient accomplie juste avant la période de sommeil. L’onde delta serait-elle
donc une période de silence sélectif, où la majorité des neurones deviennent silencieux pour ne
pas perturber une minorité de neurones qui joueraient un rôle fondamental à un moment-clef ? Pour
en avoir le coeur net, nous avons adapté la tâche de mémoire spatiale pour que les rats ne se
souviennent pas de leur expérience le lendemain. Puis nous avons provoqué des ondes delta
artificielles au bon moment, pour isoler des neurones associés aux réactivations hippocampiques, ou
quelques dizaines de millisecondes plus tard, pour isoler d’autres neurones au hasard. Résultat :
lorsque nous avons isolé les bons neurones, et seulement dans ce cas, les rats ont pu stabiliser leurs
souvenirs et ont parfaitement réussi le test le lendemain.
Ces résultats suggèrent donc une profonde révision de notre compréhension du cortex : les ondes
delta ne seraient pas des périodes de silence généralisé où le cortex devient inactif et se repose, mais
un moyen d’isoler très sélectivement des assemblées de neurones choisies, qui génèrent et
maintiennent une information cruciale entre les périodes de dialogue hippocampo-cortical et de
réorganisation des circuits corticaux, pour former des souvenirs à long terme.
Référence
Todorova R, Zugaro M. Isolated cortical computations during delta waves support memory consolidation. Science. 2019 Oct 18;366(6463):377-381. doi: 10.1126/science.aay0616.
Contact chercheur
Center for Interdisciplinary Research in Biology (CIRB), Collège de France, CNRS, INSERM, Université PSL, Paris, France. michael.zugaro@college-de-france.fr