Uncategorized
Tau et dégénérescences neurofibrillaires: toxiques OR NOT toxiques ?
Les tauopathies constituent un groupe de maladies neurodégénératives caractérisées par l’hyperphosphorylation de la protéine Tau et son accumulation sous forme d’agrégats présents dans plusieurs types cellulaires. Parmi les différent types d’agrégats intracellulaires de Tau contenus dans les neurones, les dégénérescences neurofibrillaires (DNF) sont caractéristiques des lésions retrouvées chez les patients souffrant de la maladie d’Alzheimer (MA) et d’autres tauopathies pures telles que certaines démences fronto-temporales.
Parce que le nombre de DNF chez les patients corrèle fortement avec la mort neuronale et le déclin cognitif, on a longtemps pensé que ces formes très agrégées de Tau étaient à l’origine de la neurodégénérescence. Pourtant, mort neuronale et DNF n’apparaissent pas dans les mêmes zones du cerveau chez les patients souffrant de la MA. De plus, des études électrophysiologiques ont montré que les neurones porteurs de DNF dans des modèles de souris transgéniques fonctionnent encore en réseau et survivent à l’agrégation de Tau. Ainsi, l’hypothèse que des formes oligomériques solubles de Tau ayant un état d’agrégation intermédiaire pourraient être davantage toxiques que les DNF a été émise.
Pour élucider la toxicité des DNF in vivo, nous avons généré un modèle de tauopathie, appelé modèle « pro-agrégation » chez le rat qui génère un grand nombre de DNF dans l’hippocampe. Ce modèle est basé sur l’expression simultanée de la protéine Tau humaine sauvage et d’un peptide pro-agrégation via l’utilisation de vecteurs adéno-associés (AAV). Il est caractérisé par une forte hyperphosphorylation pathologique de la protéine Tau, la localisation aberrante de la protéine dans le soma et les dendrites neuronaux et la présence de nombreuses DNF argyrophylles dès un mois après l’injection du vecteur AAV, similaires aux lésions retrouvées chez les patients. En comptant le nombre de neurones CA1 de l’hippocampe, nous avons montré que la présence de DNF ne provoque pas de mort neuronale au moins jusqu’à 3 mois après injection. A l’inverse, la surexpression de la protéine Tau humaine sauvage seule provoque une mort neuronale importante de ces mêmes neurones, ainsi qu’une forte hyperphosphorylation de Tau mais cette fois, en l’absence totale de DNF.
In vivo les agrégats matures de Tau sembleraient donc être inoffensifs pour les neurones, au moins dans un premier temps, alors que les formes solubles, sans doute oligomériques, seraient les plus toxiques. Par ailleurs, l’étude montre que le nombre de DNF reflète de manière imparfaite la sévérité de la pathologie alors que l’hyperphosphorylation de Tau sur l’épitope reconnu par l’anticorps AT8 serait un bien meilleur index de neurodégénérescence. Une implication importante de cette étude est qu’il semblerait plus approprié de développer des traceurs d’imagerie et des agents thérapeutiques capables de cibler spécifiquement ces formes solubles de Tau plutôt que les DNF.
Référence:
Potentiating tangle formation reduces acute toxicity of soluble tau species in the rat. d’Orange M, Aurégan G, Cheramy D, Gaudin-Guérif M, Lieger S, Guillermier M, Stimmer L, Joséphine C, Hérard AS, Gaillard MC, Petit F, Kiessling MC, Schmitz C, Colin M, Buée L, Panayi F, Diguet E, Brouillet E, Hantraye P, Bemelmans AP, Cambon K. Brain. 2017 Dec 14. doi: 10.1093/brain/awx342
Contact chercheuse:
Karine Cambon
CEA, DRF, Institut François Jacob, Molecular Imaging Research Center (MIRCen), F-92265 Fontenay-aux-Roses, France.
CNRS, CEA, Paris-Sud Univ., Univ. Paris-Saclay, Neurodegenerative Diseases Laboratory (UMR9199), F-92265, Fontenay-aux-Roses, France.
karine.cambon@cea.fr
Contrôler l’activité des mGluR avec des nanobodies
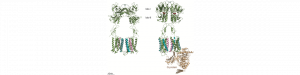
Le neurotransmetteur de la grande majorité des synapses excitatrices, le glutamate, exerce aussi des rôles modulateurs via l’activation de récepteurs couplés aux protéines G, aussi appelés les récepteurs métabotropiques du glutamate (mGluR). Ces récepteurs sont des dimères constitutifs. Huit gènes codent pour les sous-unités des mGluR, et nous avons récemment montré que ces huit sous-unités peuvent former des hétérodimères de composition définie, augmentant ainsi le nombre possible de récepteurs de huit (homodimères) à 24 (en incluant les hétérodimères possibles). Parmi ces récepteurs, ceux contenant la sous-unité mGlu2 constituent des cibles intéressantes pour le traitement de l’anxiété ou de la schizophrénie. Ainsi, de nombreuses entreprises pharmaceutiques ont développé des agonistes et des modulateurs allostériques positifs de mGlu2. Cependant, ces composés ont échoué dans les essais cliniques de phase 3 en raison d’effets secondaires ou de manque d’efficacité, qui pourraient être dus aux actions “off-target” de ces composés ou à l’existence de mGlu hétérodimériques. Dans cette étude impliquant 5 équipes françaises, dont deux de l’IGF (Pin/Rondard et Valjent) nous décrivons des nanobodies (des anticorps simple chaîne de lamas) qui agissent spécifiquement sur les homodimères mGlu2, comme des modulateurs allostériques positifs (ils augmentent les effets d’un agoniste). Ces nanobodies sont donc les premiers composés sélectifs des récepteurs mGlu2 homodimériques. Nous montrons qu’après injection dans le cerveau, l’un d’eux inhibe la consolidation de la mémoire de peur, processus dans lequel le récepteur mGlu2 homodimérique est impliqué. Cette étude montre le potentiel des nanobodies comme outils pharmacologiques mais aussi comme agents thérapeutiques potentiels pour contrôler l’activité des récepteurs dans le cerveau.
The transmitter of most excitatory synapses in the brain, glutamate, also has modulatory role through the activation of G protein-coupled receptors, the so-called metabotropic glutamate receptors (mGluRs). The mGlu receptors are mandatory dimers. Eight genes encode mGlu subunits, and we recently reported that they can assemble into heterodimers, increasing the number of possible mGlu receptors from 8 homodimers, to 24 different subtypes including the possible heterodimers. These receptors are considered as possible new targets for the treatment of various brain diseases. Among them, mGlu2 containing receptors are of interest for the treatment of anxiety and schizophrenia. As such most pharmaceutical companies developed mGlu2 receptor agonists and positive allosteric modulators, but all failed in clinical trials, mostly due to side effects or lack of efficacy. This may be partly due to the off- target effect of allosteric modulators, or the existence of mGlu2 heterodimers. In the present study, involving five research groups in France including two from the IGF (Pin/Rondard and Valjent), we identified nanobodies (single chain antibodies from lamas) specifically acting at mGlu2 homodimers, with positive allosteric properties (they enhance the action of agonist). These nanobodies are then the first highly selective positive modulators of mGlu2 homodimers. We show that injected into the brain, these nanobodies inhibit the consolidation of fear memory, implicating hippocampal mGlu2 homodimers in this process. This study reveals the great potential of nanobodies as pharmacological tools and possibly as therapeutic agents for controlling brain receptors.
Référence: Scholler P, Nevoltris D, de Bundel D, Bossi S, Moreno-Delgado D, Rovira X, Møller TC, El Moustaine D, Mathieu M, Blanc E, McLean H, Dupuis E, Mathis G, Trinquet E, Daniel H, Valjent E, Baty D, Chames P, Rondard P, Pin JP. Allosteric nanobodies uncover a role of hippocampal mGlu2 receptor homodimers in contextual fear consolidation. Nat Commun. 2017 Dec 6;8(1):1967. doi: 10.1038/s41467-017-01489-1.
Link: http://rdcu.be/AyD2
Contact chercheurs :
Institut de Génomique Fonctionnelle, CNRS, INSERM, Univ. Montpellier, F-34094, Montpellier, France.
jean-philippe.pin@igf.cnrs.fr
Philippe.Rondard@igf.cnrs.fr
Microbiote et microglies: une inégalité des sexes !
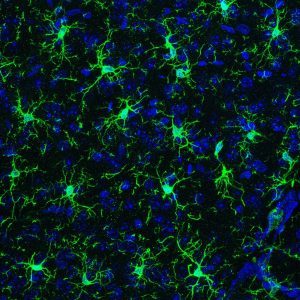
Une étude conjointe entre des chercheurs Inserm de l’IBENS (Institut de Biologie de l’Ecole Normale Supérieure) à Paris et des chercheurs du SIgN (Singapore Immunology Network, A*STAR) de Singapour montre un rôle inédit du microbiote sur des cellules immunitaires du cerveau dès le stade fœtal. Ces cellules immunitaires, les microglies, jouent un rôle clé dans le développement et le fonctionnement cérébral et sont différemment perturbées par des modifications du microbiote chez les souris mâles et femelles à différents stades de la vie. Les résultats de ces travaux sont publiés dans la revue Cell.
Les microglies sont des cellules immunitaires qui répondent à des traumatismes ou des signaux inflammatoires pour protéger le cerveau, agissant comme des senseurs capables de détecter de nombreux signaux environnementaux. Ces cellules immunitaires sont également impliquées dans différentes étapes du développement et du fonctionnement cérébral. Ainsi, des dysfonctionnements de ces cellules sont associés à un large spectre de pathologies humaines, allant des troubles neuro-développementaux jusqu’aux maladies neurodégénératives. Les microglies jouent donc un rôle crucial dans le fonctionnement normal et pathologique du cerveau, ce qui laisse suggérer qu’elles constituent une interface régulatrice entre les circuits cérébraux et l’environnement.
Pour tester cette hypothèse, Morgane Thion et Sonia Garel, chercheuses Inserm et leurs collaborateurs, ont utilisé une approche multidisciplinaire sur des modèles de souris axéniques, qui n’ont pas de microbiote (ensemble des bactéries présentes dans l’organisme) et des modèles de souris adultes traitées avec un cocktail d’antibiotiques (qui détruisent de façon aigue le microbiote). En combinant analyses génomiques globales et études histologiques, les chercheurs ont montré que les microglies sont profondément affectées par un dysfonctionnement du microbiote, dès les stades prénataux et ce, en fonction du sexe de l’animal : les microglies appartenant à des mâles semblent affectées au stade prénatal alors que les microglies issues de femelles le sont à l’âge adulte. Ce surprenant dimorphisme sexuel fait écho au fait que l’occurrence de nombreuses pathologies neurodéveloppementales est plus élevée chez les hommes alors que les maladies auto-immunes sont plutôt prévalentes chez les femmes.
Si les mécanismes impliqués et les conséquences fonctionnelles restent à découvrir, cette étude révèle un rôle clé des microglies à l’interface entre environnement et cerveau et montre que les mâles et femelles auraient des susceptibilités différentes à des altérations du microbiote. Pour les auteurs, ces éléments mériteraient maintenant d’être pris en considération au niveau clinique et ce, dès les stades fœtaux.
Réréfence: Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, Blecher R, Ulas T, Squarzoni P, Hoeffel G, Coulpier F, Siopi E, David FS, Scholz C, Shihui F, Lum J, Amoyo AA, Larbi A, Poidinger M, Buttgereit A, Lledo PM, Greter M, Chan JKY, Amit I, Beyer M, Schultze JL, Schlitzer A, Pettersson S, Ginhoux F, Garel S. Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell. 2017 Dec 21. pii: S0092-8674(17)31432-0. doi: 10.1016/j.cell.2017.11.042
Contact chercheuse:
Sonia Garel, Directrice de recherche Inserm, Institut de Biologie de l’Ecole normale supérieure (IBENS), Ecole Normale Supérieure, Paris. Tel : 06 33 49 48 14. Mel : garel@biologie.ens.fr
Une enzyme cruciale pour les neurones enfin démasquée !
L’équipe Physiopathologie du Cytosquelette de l’institut des Neurosciences de Grenoble a identifié l’enzyme responsable de la détyrosination de la tubuline qui était recherchée depuis 40 ans. Ces travaux ouvrent de nouvelles pistes pour mieux comprendre le rôle de cette modification de la tubuline dont les altérations accompagnent cancers, défauts neuronaux et maladies cardiaques. Ces résultats ont été publiés le 16 novembre 2017 dans la revue Science.
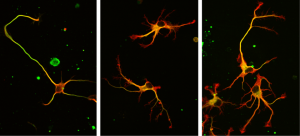
Les microtubules sont des fibres dynamiques du cytosquelette formées par l’assemblage de tubuline α et tubuline β (dimères α/β). Ils sont impliqués dans la division des cellules, leur morphologie, leur polarité, et leur migration. Ces diverses fonctions sont régulées grâce à l’existence de signaux présents à la surface de microtubules. Ces signaux, qui sont des modifications biochimiques des acides aminés des tubulines (nommés modifications post-traductionnelles parce qu’elles ont lui après la synthèse des protéines) ont lieu dans de multiples sites de la cellule et sont réalisées par diverses enzymes.
L’activité de l’une de ces enzymes a été mise en évidence pour la première fois en 1977 par des chercheurs argentins qui lui donnent le nom de TCP (Tubuline CarboxyPeptidase). Cette enzyme a comme fonction de cliver le dernier acide aminé, une tyrosine, de l’extrémité de la tubuline α. C’est la réaction de détyrosination. Une enzyme réverse, la ligase TTL (Tubuline Tyrosine Ligase), est chargée de repositionner cette tyrosine. C’est la tyrosination. Ce cycle de détyrosination/tyrosination est crucial pour la cellule et l’organisme. Une détyrosination massive (anormale) est observée dans plusieurs cancers sévères et maladies cardiaques. Il y a plusieurs années l’équipe ‘Physiopathologie du cytosquelette’ a montré que des souris n’exprimant plus la Tyrosine Ligase (TTL) meurent à la naissance parce que leur cerveau ne se développe pas correctement.
Dans ce travail récent, pour identifier la TCP recherchée depuis quatre décennies, l’équipe a suivi son activité, utilisé des techniques classiques de biochimie et collaboré avec des chimistes de l’Université de Stanford qui ont développé un inhibiteur ‘suicide’ de l’enzyme. Cette molécule a été utilisée comme hameçon pour ‘pêcher’ l’enzyme convoitée peu abondante dans les tissus. C’est finalement deux enzymes qui ont été découvertes! Ces dernières, dénommées VASH1 et VASH2 (vasohibin 1 et 2), étaient déjà connues des scientifiques mais sans savoir qu’il s’agissait d’enzymes en lien avec le cytosquelette. A la condition d’être associées à une protéine partenaire appelée SVBP, VASH1 et VASH2 sont capables de détyrosiner la tubuline α. La suppression de leur expression (ou de celle de leur partenaire SVBP) dans les neurones engendre une très forte diminution du taux de détyrosination de la tubuline α, ainsi que des anomalies dans la morphologie des neurones. La différenciation des neurones est retardée. En collaboration avec l’équipe “Progéniteurs Neuraux et Pathologies Cérébrales”, les chercheurs ont également montré que ces enzymes sont impliquées lors du développement du cortex cérébral.
Dorénavant, les chercheurs espèrent qu’en modulant l’efficacité de la TCP et en améliorant la connaissance du cycle détyrosination/tyrosination, il sera possible de progresser dans la compréhension de ses fonctions cérébrales et cardiaques, et de mieux lutter contre certaines maladies.
Référence:
Vasohibins/SVBP are tubulin carboxypeptidases (TCP) that regulate neuron differentiation.
Aillaud C, Bosc C, Peris L, Bosson A, Heemeryck P, Van Dijk J, Le Friec J, Boulan B, Vossier F, Sanman LE, Syed S, Amara N, Couté Y, Lafanechère L, Denarier E, Delphin C, Pelletier L, Humbert S, Bogyo M, Andrieux A, Rogowski K, Moutin MJ. Science. 2017 Nov 16. doi: 10.1126/science.aao4165.
Contact:
Marie-Jo Moutin
Grenoble Institut des Neurosciences
Equipe Physiopathologie du Cytosquelette
Inserm U1216 – UGA
Chemin Fortuné Ferrini
Bâtiment Edmond J. Safra
38700 La Tronche
Tél : 04.56.52.05.35
Mail : moutinm@univ-grenoble-alpes.fr
Construire des ponts à travers les Neurosciences
Un article commun a dernièrement été publié dans Neuron, co-signé par des neuroscientifiques reconnus appartenant à un très large panel de spécialités et de pays : Allemagne, France, Egypte, Israël, Espagne, Suisse, Etats-Unis d’Amérique, Turquie, Pakistan, Italie …
L’angle de l’article est celui des Neurosciences présentées comme étant une discipline parfaitement adaptée à la reconnexion de l’Occident et du Moyen-Orient. Après tout, la quête pour comprendre le cerveau touche à la nature même de ce que nous sommes en tant qu’individus et en tant qu’espèce. Le développement des théories et pratiques liées au cerveau a une longue tradition au Moyen-Orient, illustrée par la vision moniste de l’esprit et du corps (par opposition au dualisme cartésien) et l’importance d’une approche holistique dans le traitement des maladies psychiatriques. Contrairement à d’autres parties du monde, où les malades mentaux étaient ostracisés et possédés par les démons, le Moyen-Orient médiéval avait un fort intérêt intellectuel dans la prise en charge des troubles psychiatriques.
L’initiative TSB poursuit une stratégie qui intègre des objectifs à court et à long terme. À court terme, l’objectif est de faciliter la collaboration et la libre circulation entre les instituts de recherche existants en établissant des programmes d’échange d’étudiants et de chercheurs, en partageant des subventions de recherche collaborative et en organisant des symposiums scientifiques entre les laboratoires participants. L’objectif à long terme est la création de “Twin-Institutes TSB”, un institut de recherche étant situé dans un pays occidental et l’autre dans un pays du Moyen-Orient. Les deux établissements collaboreront intensément grâce à une infrastructure encourageant le dialogue interculturel et disposera d’un département dédié aux ressources humaines, spécialisé dans les problèmes spécifiques auxquels pourraient faire face les différents chercheurs participant aux collaborations Est-Ouest, et accordera une attention particulière à la communication des résultats de ces études au public. Il existe un grand potentiel inexploité dans les pays du Moyen-Orient. L’initiative TSB soutiendra fortement la formation locale et l’intégration des scientifiques nés dans les pays du Moyen-Orient, ayant reçu une partie de leur éducation ailleurs et souhaitant retourner dans leur pays d’origine pour y établir et nourrir une culture scientifique accomplie.
Nous nous devons de continuer à transformer le dialogue interculturel et trouver des dénominateurs communs. Avec toute une variété de systèmes de croyances humaines parfois très différente, la santé et la connaissance sont les deux choses que chaque personne chérit et mérite d’acquérir et de conserver. L’initiative TSB soutient que les progrès scientifiques seront absolument cruciaux pour la consolidation de la paix dans le monde. En biologie, la diversité permet une adaptation réussie, qu’il s’agisse de cellules, d’organismes ou de populations entières. Par analogie, la diversité culturelle et géographique devrait nous permettre d’aborder plus efficacement les problèmes les plus difficiles concernant le cerveau, tels que le mystère de la conscience et l’impact dévastateur des troubles psychiatriques et neurologiques. Le TSB vise à créer une nouvelle dimension de la recherche collaborative axée sur l’avancement de la science cérébrale fondamentale et translationnelle avec l’espoir que cette initiative ouvrira un nouveau chapitre dans l’échange scientifique Est-Ouest et contribuera profondément à l’harmonie interculturelle et au progrès scientifique.
Référence :
Lissek T, Adams M, Adelman J, Ahissar E, Akaaboune M, Akil H, al’Absi M, Arain F, Arango-Lasprilla JC, Atasoy D, Avila J, Badawi A, Bading H, Baig AM, Baleriola J, Belmonte C, Bertocchi I, Betz H, Blakemore C, Blanke O, Boehm-Sturm P, Bonhoeffer T, Bonifazi P, Brose N, Campolongo P, Celikel T, Chang CC, Chang TY, Citri A, Cline HT, Cortes JM, Cullen K, Dean K, Delgado-Garcia JM, Desroches M, Disterhoft JF, Dowling JE, Draguhn A, El-Khamisy SF, El Manira A, Enam SA, Encinas JM, Erramuzpe A, Esteban JA, Fariñas I, Fischer E, Fukunaga I, Gabilondo I, Ganten D, Gidon A, Gomez-Esteban JC, Greengard P, Grinevich V, Gruart A, Guillemin R, Hariri AR, Hassan B, Häusser M, Hayashi Y, Hussain NK, Jabbar AA, Jaber M, Jahn R, Janahi EM, Kabbaj M, Kettenmann H, Kindt M, Knafo S, Köhr G, Komai S, Krugers H, Kuhn B, Ghazal NL, Larkum ME, London M, Lutz B, Matute C, Martinez-Millan L, Maroun M, McGaugh J, Moustafa AA, Nasim A, Nave KA, Neher E, Nikolich K, Outeiro T, Palmer LM, Penagarikano O, Perez-Otano I, Pfaff DW, Poucet B, Rahman AU, Ramos-Cabrer P, Rashidy-Pour A, Roberts RJ, Rodrigues S, Sanes JR, Schaefer AT, Segal M, Segev I, Shafqat S, Siddiqui NA, Soreq H, Soriano-García E, Spanagel R, Sprengel R, Stuart G, Südhof TC, Tønnesen J, Treviño M, Uthman BM, Venter JC, Verkhratsky A, Weiss C, Wiesel TN, Yaksi E, Yizhar O, Young LJ, Young P, Zawia NH, Zugaza JL, Hasan MT.
Building Bridges through Science. Neuron. 2017 Nov 15;96(4):730-735. doi: 10.1016/j.neuron.2017.09.028.
Contact :
Mohamed Jaber
Inserm UMR_S 10184, Université de Poitiers
Courriel
Figure issue de la publication dans Neuron montrant l’objectif commun de l’initiative “science bridge”.
UN GÈNE ASSOCIÉ À LA SCHIZOPHRÉNIE CONTRÔLE L’ACTIVITÉ DES NEURONES DOPAMINERGIQUES

Une étude menée par Bertrand Lambolez et Ludovic Tricoire de l’unité Neuroscience Paris-Seine, parue le 11 juillet 2017 dans la revue Molecular Psychiatry, réconcilie deux hypothèses qui expliquent l’origine de la schizophrénie. Leurs travaux portent sur la protéine membranaire GluD1, codée par le gène Grid1, dont les mutations sont associées à des cas de schizophrénie et de troubles bipolaires.
La schizophrénie est une maladie psychiatrique caractérisée par plusieurs symptômes : des délires et hallucinations, et/ou un retrait social et des difficultés cognitives. Deux hypothèses s’affrontent pour en expliquer l’origine, impliquant deux neurotransmetteurs majeurs du cerveau, le glutamate et la dopamine. L’hypothèse dopaminergique postule qu’un dysfonctionnement des neurones qui libèrent la dopamine est en cause, tandis que l’hypothèse glutamatergique est fondée sur le fait qu’un blocage des récepteurs au glutamate mime certains symptômes de la maladie.
Le glutamate, principal neurotransmetteur excitateur du cerveau, contrôle l’activité des neurones par le biais de deux types de récepteurs : les récepteurs métabotropes (mGlu), et les ionotropes (iGlu) qui forment un canal ionique. Parmi ces derniers, le récepteur delta GluD1 a longtemps intrigué les neurobiologistes : bien que structurellement semblable aux autres récepteurs ionotropes, il ne répond pas au glutamate. Pendant des décennies, ce récepteur a donc été considéré comme « orphelin », aucune molécule endogène ne permettant l’ouverture de son canal ionique.
Dans cette étude, les chercheurs ont étudié le rôle de GluD1 dans la neurotransmission glutamatergique sur les neurones dopaminergiques (DA) du cerveau. Ils ont découvert le mode d’activation original de GluD1 : il répond indirectement glutamate, via l’activation en amont de récepteurs métabotropes mGlu 1/5 exprimés par les neurones DA. Ainsi, l’activation de mGlu 1/5 déclenche une excitation des neurones DA, supprimée par une mutation qui bloque le canal ionique de GluD1, ou par délétion du gène Grid1.
L’activité des neurones dopaminergiques in vivo présente deux profils distincts : soit une activité électrique régulière à basse fréquence, soit des bouffées d’activité électrique à haute fréquence. Ce second mode de fonctionnement est associé à un pic de libération de dopamine dans le cerveau. Des enregistrements in vivo ont révélé que les souris dont le récepteur GluD1 est muté ou absent ne présentent plus d’activité haute fréquence, démontrant la relation entre GluD1 et l’activité physiologique des neurones DA.
Ces travaux fournissent des éléments permettant de réconcilier les hypothèses dopaminergique et glutamatergique de la schizophrénie en révélant une interconnexion entre les deux types de neurotransmissions au sein même des neurones DA. En montrant que l’inactivation génétique de GluD1 abolit l’activité haute fréquence des neurones DA, ces travaux expliquent comment une altération du gène Grid1 impliqué dans la transmission glutamatergique peut mener à un dysfonctionnement du système dopaminergique. Autre résultat important : en identifiant un mode d’activation physiologique de GluD1, cette étude lève l’état « orphelin » de ce récepteur.
Références :
Benamer N, Marti F, Lujan R, Hepp R, Aubier TG, Dupin AAM, Frébourg G, Pons S, Maskos U, Faure P, Hay YA, Lambolez B, Tricoire L. GluD1, linked to schizophrenia, controls the burst firing of dopamine neurons. Mol Psychiatry. 2017 Jul 11. doi: 10.1038/mp.2017.137.
Contact chercheurs:
Sorbonne Universités, UPMC Univ Paris 06 UM119, Centre National de la Recherche Scientifique (CNRS) UMR8246, Institut National de la Santé et de la Recherche Médicale (INSERM) UMR-S1130, Neuroscience Paris Seine, Institut de Biologie Paris-Seine, Paris, France
Les récepteurs synaptiques mGluR5 en mouvement : fauteurs de troubles dans le syndrome du X fragile
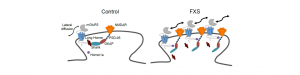
Le syndrome du X fragile (SXF) est la forme héréditaire la plus courante de déficience intellectuelle et une cause fréquente de troubles du spectre autistique (TSA). Le sous-type 5 du récepteur métabotropique au glutamate (mGluR5) est crucial dans la physiopathologie du SXF. Cependant, son dysfonctionnement au niveau subcellulaire par rapport aux phénotypes synaptiques et cognitifs du SXF est largement inexploré. mGluR5 s’associe avec un certain nombre de protéines synaptiques et ces interactions influencent fortement les propriétés dynamiques du récepteur à la synapse, ainsi que la fonction d’autres récepteurs membranaires.
Des travaux antérieurs ont démontré que l’interaction entre mGluR5 et les formes longues de la protéine d’échafaudage, Homer, est réduite chez les souris déficientes pour le gène Fmr1 (modèle murin pour SXF) et que ces altérations sont causées par une surexpression de Homer1a, l’une des isoformes d’Homer. Les conséquences de cette interaction altérée pour la dynamique des récepteurs n’avaient cependant pas été étudiées auparavant.
Dans cette étude, nous avons sondé les conséquences de la perturbation du complexe contenant mGluR5/Homer pour la mobilité de mGluR5 à la surface cellulaire, pour la fonction synaptique du récepteur N-méthyl-D-aspartate (NMDAR) et pour les phénotypes comportementaux chez les souris Fmr1 -/y.
Par le suivi d’une molécule unique, nous avons trouvé que mGluR5 était significativement plus mobile aux synapses des neurones Fmr1-/y de l’hippocampe, provoquant une augmentation de la co-localisation à la surface synaptique de mGluR5 et NMDAR. Ceci est corrélé avec une diminution de l’amplitude des courants synaptiques liée au NMDAR, à l’absence de leur dépression à long terme activée par mGluR5 et à des déficits cognitifs NMDAR / hippocampe. Ces phénomènes synaptiques et comportementaux ont été inversés en inhibant l’expression de l’isoforme courte Homer1a, dans l’hippocampe des souris Fmr1 -/y.
Notre étude fournit un important lien mécanistique entre les changements dans la dynamique de mGluR5 aux sites synaptiques et les phénotypes pathologiques du SXF. Ces découvertes sont susceptibles d’avoir des conséquences importantes pour le futur développement d’agents / stratégies thérapeutiques du SXF.
En effet, mGluR5 et NMDAR ont été proposés comme cibles thérapeutiques dans le SXF et le lien entre ces deux récepteurs devrait être pris en compte lors de la prédiction des résultats de thérapies unique ou combinée. De plus, la correction de l’équilibre altéré entre mGLluR5/Homer et mGluR5/NMDAR pourrait constituer une alternative thérapeutique prometteuse pour le développement de nouveaux agents thérapeutiques pour le traitement du SXF et des TSA.
Référence :
Aloisi E, Le Corf K, Dupuis J, Zhang P, Ginger M, Labrousse V, Spatuzza M, Georg Haberl M, Costa L, Shigemoto R, Tappe-Theodor A, Drago F, Vincenzo Piazza P, Mulle C, Groc L, Ciranna L, Catania MV, Frick A. Altered surface mGluR5 dynamics provoke synaptic NMDAR dysfunction and cognitive defects in Fmr1 knockout mice. Nat Commun. 2017 Oct 24;8(1):1103. doi: 10.1038/s41467-017-01191-2.
Contact :
Andreas Frick, Neurocentre Magendie, INSERM U1215, 146 rue Léo Saignat, 33077 Bordeaux Cedex
Courriel
Fragile X syndrome (FXS) is the most common inherited form of intellectual disability and a frequent cause of autism spectrum disorder (ASD). Metabotropic glutamate receptor subtype 5 (mGluR5) is crucially implicated in the pathophysiology of FXS. However, its dysfunction at the sub-cellular level in relation to the synaptic and cognitive phenotypes in FXS is largely unexplored. mGluR5 associates with a number of synaptic proteins and these interactions strongly influence the dynamic properties of the receptor at the synapse, as well as the function of other membrane receptors. Previous work has demonstrated that the interaction between mGluR5 and long forms of the scaffolding protein, Homer, is reduced in Fmr1 knockout mice (the mouse model for FXS) and that these alterations are caused by an overexpression of the short, activity-regulated Homer isoform, Homer 1a. The consequences of this altered interaction for receptor dynamics had not, however, been previously investigated.
In this study, we probed the consequences of the aforementioned mGluR5/Homer scaffold disruption for mGluR5 cell-surface mobility, synaptic N-methyl-D-aspartate receptor (NMDAR) function, and behavioral phenotypes in the Fmr1 knockout (KO) mouse. Using single-molecule tracking, we found that individual mGluR5 was significantly more mobile at synapses in hippocampal Fmr1KO neurons, causing an increased synaptic surface co-clustering of mGluR5 and NMDAR. This correlated with reduced amplitude of synaptic NMDAR currents, a lack of their mGluR5-activated long-term depression, and NMDAR/hippocampus dependent cognitive deficits. Crucially, these synaptic and behavioral phenomena were reversed by knocking down the short, activity-related Homer1a in the hippocampus of Fmr1 KO mice. Our study provides an important mechanistic link between changes in mGluR5 dynamics at synaptic sites and pathological phenotypes of FXS. These findings are likely to have important consequences for the future development of therapeutic agents/strategies for the treatment of FXS. In particular, both mGluR5 and NMDAR have been proposed as targets for therapeutic intervention in FXS and this altered crosstalk between these two receptors should be taken into consideration when predicting the outcome of single or combined therapies. In addition, correcting the altered balance between GluR5/Homer and mGluR5/NMDAR might provide a promising alternative for the development of novel therapeutic agents for the treatment of FXS and ASD.
Un rôle pour la sérotonine dans la motivation et l’effort !

Les neuromodulateurs modifient le traitement de l’information dans les réseaux de neurones où ils sont naturellement libérés. La sérotonine est un neuromodulateur dont le rôle est particulièrement complexe: elle est libérée dans de très nombreuses régions cérébrales, où elle interagit avec de nombreux récepteurs, et produit des effets très variés sur le comportement. Elle est par exemple impliquée dans la régulation de l’humeur, de l’anxiété, de l’impulsivité et de l’apprentissage. Il s’agit à ce titre d’une cible thérapeutique de premier plan.
La perte de motivation est un symptôme clé de la dépression mais le rôle de la sérotonine dans la régulation de la motivation reste actuellement très controversé. Des chercheurs français de l’Institut du Cerveau et de la Moelle épinière à Paris, de l’hôpital Saint Anne, associés à des collègues des Universités d’Oxford et de Manchester ont récemment publié les résultats d’une étude visant à clarifier le rôle de la sérotonine dans la régulation de la motivation.
Dans cette étude, réalisée chez des sujets sains, la motivation a été « opérationalisée » comme la capacité à produire des efforts soutenus dans lequel l’effort produit par le participant était récompensée par un gain monétaire. L’étude visait à évaluer si la sérotonine était susceptible de modifier la capacité à produire des efforts soit en exacerbant la perception du coût (fatigue, douleur, … ) soit en diminuant l’attrait des bénéfices associés à l’action. Une modification de la durée des efforts dans cette tâche pouvait donc être le résultat d’une modification de la sensibilité au coût ou au bénéfice de l’effort. Un point clé de l’étude est que les chercheurs proposent un modèle mathématique du comportement dans cette tâche qui permet de distinguer ces deux scénarios. Les résultats montrent que l’inhibition de la recapture de sérotonine améliore la performance dans cette tâche en diminuant spécifiquement le coût ressenti de l’effort : les participants recevant le traitement produisaient des efforts plus longs, et ils obtenaient ainsi des gains supérieurs à ceux recevant le placebo.
La sérotonine pourrait ainsi modifier les coûts d’une façon plus générale, qu’ils soient associés à un effort comme dans cette étude, ou à des délais ou même des punitions infligées dans de précédentes tâches de décision. Ce rôle général de la sérotonine dans le cerveau pourrait expliquer les nombreux effets déjà observés sur la régulation et la motivation du comportement.
Référence : Meyniel, F., Goodwin, G.M., Deakin, J.W., Klinge, C., MacFadyen, C., Milligan, H., Mullings, E., Pessiglione, M. and Gaillard, R., 2016. A specific role for serotonin in overcoming effort cost. Elife, 5, p.e17282.
Contact : Florent Meyniel – Chargé de recherche CEA, NeuroSpin, CEA-Saclay F-91191 Gif sur Yvette Cedex FRANCE
41st Annual Meeting of Japan Neuroscience Society (Neuroscience 2018)-Call for Travel Award Application
The 41st Annual Meeting of Japan Neuroscience Society (Neuroscience 2018) will be held at Kobe Convention Center in Hyogo, Japan, from July 26th to 29th, 2018.
Neuroscience 2018 offers Travel Awards to enthusiastic leading non-Japanese neuroscientists traveling from abroad to present high quality papers at the meeting. Applications of young principal investigators and senior postdoctoral fellows are especially encouraged. The funding for travel will be between 40,000-150,000 JPY per award depending on the travel distance. The awardees will also be exempt from the registration fee of the meeting. Those from Asian-Pacific countries are particularly encouraged.
Please be advised that persons from the following countries must apply for the Travel Award through their home Neuroscience Affiliations: China, South Korea, India, and Iran.
– The Japan Neuroscience Society (JNS) was founded in 1974. Since then, the membership has increased steadily. With more than 6,300 members in 2017, JNS is now a representative academic society in neuroscience in Japan.
– Neuroscience 2018 under the theme of “New Horizon of Neuroscience” provides a unique opportunity to forge closer links between researchers in different fields and to develop next-generation neuroscientists endowed with a wide perspective.
– Top international and domestic researchers will take this as an opportunity to elucidate the mechanisms of the brain and mind through promotion of a wide range of neuroscience research fields, across clinical medicine and psychology, and to the molecular and cellular biological levels.
– We will also focus our efforts on promoting interaction with relevant professional neuroscience societies in neighboring Asia-Pacific countries, the United States and Europe by organizing joint symposia and providing a financial assistance program to cover travel costs.
– All travel award winners are invited to the International Exchange Meeting for Young Researchers and a get-together party to be held on July 25th, 2018 to facilitate interaction among young researchers from abroad and Japan. In addition, they will be invited to a reception on DAY 2 of the Annual Meeting.
– The Annual Meeting aims at fostering young scientists and promoting collaboration across the globe and various scientific fields including clinical neuroscience. We expect approximately 4,000 participants, including 400 people from overseas.
– The Meeting will be held at Kobe Convention Center, Hyogo.
Application deadline: January 9th, 2018.
Un modèle animal inédit pour accélérer la lutte contre la maladie d’Alzheimer !
La maladie d’Alzheimer reste aujourd’hui incurable. Parmi les obstacles rencontrés pour développer des traitements efficaces figure l’impossibilité de « cibler » la maladie avant un stade avancé. Or, les caractéristiques biologiques de la maladie d’Alzheimer apparaissent au moins vingt ans avant la manifestation des symptômes cliniques (perte de mémoire, troubles émotionnels, etc.). Il faudra donc comprendre cette phase silencieuse (ou infra-clinique) pour espérer pouvoir soigner les patients alors que les atteintes cérébrales sont encore réversibles. Jusqu’à présent, il n’existait pas de modèles in vitro ou animaux qui permettent d’étudier cette longue période précédant l’apparition de symptômes cognitifs. La collaboration entre des équipes de chercheurs du département MIRCen du CEA, de l’Inserm, des universités Paris-Sud et Paris-Descartes et du CNRS a abouti au développement d’une technologie disruptive qui permet d’induire chez des rats adultes une « maladie d’Alzheimer » présentant les deux types de lésions et mimant de manière fidèle la progression de celle-ci dès ses premières phases de développement.
Dans l’article publié dans le journal Cerebral Cortex, le modèle, baptisé AgenT, est induit chez le rat à l’âge adulte par co-transfert stable des gènes humains mutés APP et PS1 à l’aide de Virus Adéno-Associés (AAV) dans l’hippocampe des animaux. Cette approche a permis la production localisée des protéines APP et PS1 dans un nombre très réduit de neurones. Ces neurones vont produire des peptides Aß42 qui vont alors diffuser dans l’ensemble du tissu hippocampique. La grande majorité du tissu hippocampique ne présente ainsi pas de modification génétique, ce qui en fait un modèle pertinent pour les formes non génétiques de la maladie qui représentent plus de 95% des cas. Sa pertinence physiopathologique a été validée en le comparant à des échantillons post-mortem de patients. La concentration des peptides Aß42 va progressivement augmenter pour atteindre en fin de vie de l’animal des concentrations comparables à celles mesurées dans l’hippocampe des patients. Une hyper-phosphorylation de la protéine Tau endogène va alors graduellement se mettre en place, simultanément les capacités de mémoire vont progressivement décliner, reproduisant la cinétique de progression de la pathologie humaine. Les plaques amyloïdes et l’angiopathie amyloïde cérébrale ne se développent qu’en fin de vie de l’animal. Des agrégats intra-neuronaux de protéine Tau hyper-phosphorylée confirment un engagement complet de la pathologie Tau.
Le fait de disposer de rongeurs modélisant de manière pertinente les stades cliniquement silencieux de la maladie va ainsi permettre 1/ d’étudier la phase précoce de la maladie, phase où le développement de celle-ci semble encore réversible, 2/ d’évaluer l’efficacité des candidats médicaments en caractérisant leurs effets sur les deux voies pathologiques (amyloïde et/ou Tau) et 3/ de définir des marqueurs sanguins spécifiques des stades précoces de la maladie, qui pourraient permettre un diagnostic dès 50 ans chez l’Homme.
Références :
Audrain M1,2,3, Souchet B1,3,4, Alves S1,3, Fol R1,2,3, Viode A5, Haddjeri A6, Tada S1,3, Orefice NS1,3, Joséphine C3,7, Bemelmans AP3,7, Delzescaux T3,7, Déglon N8,9, Hantraye P3,7,10, Akwa Y11, Becher F5, Billard JM6, Potier B6, Dutar P6, Cartier N1,3, Braudeau J1,3.
βAPP Processing Drives Gradual Tau Pathology in an Age-Dependent Amyloid Rat Model of Alzheimer’s Disease.
Cerebral Cortex. 2017. DOI 10.1093/cercor/bhx260
1 INSERM UMR1169, Université Paris-Sud, Université Paris-Saclay, Orsay 94100, France.
2 Université Paris Descartes, Paris, France.
3 CEA, DRF, Institut Francois Jacob, MIRCen, Fontenay-aux-Roses 92265, France.
4 Université Paris Saclay, Paris, France.
5 CEA, Institut Frédéric Joliot, Service de Pharmacologie et d’Immunoanalyse, 91191, Gif-sur-Yvette, France.
6 INSERM UMR894, Centre de Psychiatrie et Neurosciences, Université Paris Descartes, Sorbonne Paris Cité, Paris, France.
7 CNRS UMR9199, Fontenay-aux-Roses 92265, Université Paris-Sud, Université Paris-Saclay, Orsay 94100, France.
8 Department of Clinical Neurosciences, Laboratory of Cellular and Molecular Neurotherapies, Lausanne University Hospital, Lausanne, Switzerland.
9 Neuroscience Research Center, Laboratory of Cellular and Molecular Neurotherapies, Lausanne University Hospital, Lausanne, Switzerland.
10 INSERM UMS27, Fontenay-aux-Roses 92265, Université Paris-Sud, Université Paris-Saclay, Orsay 94100, France.
11 INSERM U1195 and Université Paris Sud and Université Paris-Saclay, 80 rue du général Leclerc, 94276 Le Kremlin-Bicêtre, France.
Communiqué de presse :
Contact :
Jérôme Braudeau, Ph.D.
AgenT, CEA Fontenay aux Roses, Bât 61, Bureau 125, 18 route du panorama, 92260 Fontenay-Aux-Roses, France