Uncategorized
Circuits thalamocorticaux de la prise de décision
La capacité à prendre une décision adaptée dans un environnement changeant fait intervenir de multiples régions cérébrales interconnectées. Le cortex préfrontal est l’un des sites primordiaux mais ces dernières années ont vu apparaître le rôle important des régions thalamiques. Actuellement, l’implication de ces circuits thalamocorticaux dans les fonctions cognitives est reconnue comme déterminante, particulièrement en ce qui concerne les liens entre le cortex préfrontal (PFC) et le thalamus médiodorsal (MD). Le cortex préfrontal se caractérise néanmoins par une importante hétérogénéité anatomique, à laquelle fait écho la diversité des projections thalamocorticales issues du MD : différentes populations neuronales thalamiques innervent les différentes régions préfrontales.
Récemment, l’équipe Décision et Adaptation de l’INCIA (Institut de Neurosciences Cognitives et Intégratives d’Aquitaine, UMR 5287, Bordeaux) a pu établir que les connexions entre le mPFC et le MD sont essentielles pour la capacité à prendre une décision adaptée en usant d’approches pharmacogénétiques avancées chez le rat. Ces résultats lèvent un voile partiel sur la contribution fonctionnelle des connections entre cortex préfrontal et thalamus en se focalisant spécifiquement sur le mur médian du cortex préfrontal. Néanmoins, le cortex orbitofrontal (OFC) fait également l’objet d’une innervation importante du MD et d’une autre région thalamique largement méconnue, le thalamus submédian.
Pour donner un éclairage plus complet sur le rôle fonctionnel de ces circuits, l’équipe a donc déconnecté l’OFC de l’une et l’autre de ses afférences thalamiques et évalué l’impact de ces manipulations sur la capacité à prendre une décision fondée sur la valeur courante du but chez le rat. Ceci est classiquement effectué en utilisant une procédure de dévaluation spécifique du but, après un apprentissage instrumental initial dans lequel les animaux apprennent que l’appui sur deux leviers différents permet d’obtenir une récompense alimentaire spécifique. Après cette phase, la procédure de dévaluation consiste à fournir à l’animal l’une de ces deux récompenses à volonté, et à procéder à un test de choix entre les deux leviers immédiatement après. Lors de ce test, les leviers sont inactifs et les choix des animaux sont donc fondés uniquement sur leur représentation de la valeur courante de la récompense : des animaux « normaux » répondent majoritairement sur le levier correspondant à la récompense non dévaluée. De façon intéressante, les déconnections entre cortex orbitofrontal et thalamus médiodorsal ou submédian n’ont pas produit d’effet, suggérant un rôle spécifique du circuit mPFC-MD (Alcaraz et al., 2018). Néanmoins, après une phase d’apprentissage instrumental supplémentaire dans lequel la flexibilité cognitive des animaux est sollicitée, en inversant les contingences entre levier et nourriture qui lui est associée, un résultat bien différent apparaît. Alors que la déconnection de l’OFC et du MD est toujours sans effet, déconnecter l’OFC de son autre afférence thalamique, le thalamus submédian, produit un déficit spécifique lors du choix, suggérant que ces animaux ne sont pas capables de s’adapter au changement de contingences.
Ce résultat fait écho à une démonstration similaire de l’équipe ayant préalablement établi un rôle du thalamus submédian dans la capacité à mettre à jour les contingences Pavloviennes (Alcaraz et al., 2015). De façon plus générale, pris dans leur ensemble, ces études soulignent que différents circuits thalamocorticaux semblent coopérer pour assurer le caractère flexible de l’action dirigée vers un but. Un enjeu important pour les années à venir sera de déterminer les mécanismes par lesquels les échanges fonctionnels entre ces circuits peuvent opérer.
Référence
A thalamocortical circuit for updating action-outcome associations (2019) Fresno V, Parkes SL, Faugère A, Coutureau E*, Wolff M*. *Contributed equally.
Elife. 2019 Apr 23;8. pii: e46187. doi: 10.7554/eLife.46187.
Pour aller plus loin
Thalamocortical and corticothalamic pathways differentially contribute to goal-directed behaviors in the rat (2018) Alcaraz F, Fresno V, Marchand AR, Kremer EJ, Coutureau E, Wolff M.
Elife. 2018 Feb 6;7. pii: e32517. doi: 10.7554/eLife.32517.
https://lejournal.cnrs.fr/nos-blogs/aux-frontieres-du-cerveau/comment-le-cerveau-decide
http://archives.cnrs.fr/presse/article/4227
Contact chercheurs
Equipe Décision et Adaptation, Institut de Neurosciences Cognitives et Intégratives d’Aquitaine (INCIA), UMR5287, Bordeaux
Effet paradoxal des antidéprésseurs pendant le développement : une cible inattendue dans le cortex préfrontal
Les inhibiteurs de la recapture de la sérotonine (IRS) sont les antidépresseurs les plus prescrits dans la prise en charge des états dépressifs et anxieux. Leur succès tient en particulier au fait qu’ils ont peu d’effets secondaires. Cependant il pourrait en être autrement pendant le développement. C’est tout au moins ce que démontrent les études précliniques chez le rongeur, révélant un effet paradoxal des IRS quand ils sont administrés pendant le développement. En effet, pendant une période critique du développement postnatal, les IRS favorisent l’émergence de symptomes anxieux et dépréssifs chez l’adulte. Les mécanismes de cet effet paradoxal sont mal connus.
Une publication récente de l’équipe du Dr Gaspar à l’institut du Fer à Moulin (Inserm, Sorbonne Université) apporte un éclairage nouveau sur cette question. Mariano Soiza-Reilly et collaborateurs viennent de montrer que les IRS perturbent le développement des circuits reliant le cortex préfrontal au raphe. Ce sont des circuits cruciaux dans la réponse au stress notamment pour modérer les réponses anxieuses. Soiza-Reilly et al. montrent que le développement de ces circuits préfrontaux se poursuit pendant les premières semaines de vie postnatale chez le rongeur, et qu’il est durablement modifié par les IRS, administrés pendant cette même période.
Les chercheurs ont d’abord fait l’observation inattendue d’une expression transitoire du transporteur de la sérotonine (SERT) dans une sous-population de neurones pyramidaux du cortex prefrontal de la souris. Cette observation prolonge des observations antérieures de l’équipe montrant que l’expression du SERT est plus large pendant le développement que chez l’adulte, s’étendant en particulier en dehors des neurones sértoninergiques du raphe. Dans la présente étude, le marquage génétique des neurones SERT+ préfrontaux a permis leur identification transcriptomique et anatomique précises. Il s’agit de neurones pyramidaux, glutamatergiques dont les projections sous-corticales ciblent le thalamus et différents noyaux du tronc cérébral, le raphe en particulier. Ils forment des synapses excitatrices sur les neurones sérotoninergiques et Gabaèrgiques du raphe. Utilisant des approches combinées de pharmacologie et de génétique, les chercheurs ont ensuite pu démontrer que l’invalidation de SERT spécifiquement dans le cortex pendant une période critique, était nécessaire et suffisante pour induire une exubérance de synapses excitatrices dans différentes cibles sous corticales (thalamus et raphe). Pour éclairer le lien de ces circuits avec le comportement anxieux et dépressif provoqué par l’administration développementale d’IRS les auteurs ont utilisé des approches pharmacogénétiques. Celles ci ont permis de montrer que les neurones SERT+ du cortex préfrontal modulent effectivement les réponses au stress dans des tests classiques d’anxiété et de dépréssion (test de conflit, nage forcée).
Au total, cette étude montre clairement que les IRS ont des cibles cellulaires différentes pendant le développement et chez l’adulte. L’expression transitoire de SERT dans une sous-population de neuones du CPF, permet le contrôle local des taux de 5HT et régule ainsi finement la synaptogénèse. Une des question posée par cette observation est de déterminer quels récepteurs 5-HT sont impliqués dans ces effets sur la synaptogénèse. Une autre question importante est de savoir si ces données sont transposables au développement humain. Certaines données suggèrent que c’est bien le cas. Ainsi, l’analyse transcriptome du cerveau de foetus humain (Allen Brain Atlas) indique que le SERT est bien exprimé de manière transitoire dans le cortex préfrontal chez l’homme et des observations morphologiques faites dans l’équipe montrent un marquage des axones corticaux frontaux dans des cerveaux d’embryons humains de 11 semaines. Il est donc probable que des phénomènes analogues existent chez l’homme mais une echelle temporelle différente du développement.
Références
Soiza-Reilly M., Meye FJ. ,Olusakin O , Telley L, Petit E, Chen X, Mameli M, Jabaudon D, Sze J-Y, Gaspar P. SSRIs target prefrontal-raphe circuits during development to modulate synaptic connectivity and emotional behavior ; Mol Psychiatry. 2019 Jan 10. doi: 10.1038/s41380-019-0349-9.
Contact chercheur
Institut du Fer à Moulin, Paris, France
Un point clé de la théorie de l’apprentissage démontré grâce à l’optogénétique

Le cerveau apprend de manière constante pour nous permettre d’améliorer nos actions en fonction de nos expériences. Plusieurs théories visent à rendre compte de cette propriété fondamentale, l’une des plus populaires étant l’apprentissage par renforcement, utilisée aussi en intelligence artificielle. Cette théorie postule que l’apprentissage émerge grâce à un renforcement spécifique des connections entre les neurones qui sont actifs durant un événement, une action ou une suite d’événements et d’actions menant à une récompense. Un des point clé de cette théorie est que plus les neurones impliqués sont actifs, plus le renforcement des connections est rapide et solide. Ainsi les événements qui activent le plus fortement notre cerveau devraient être appris de manière prioritaire par rapport à d’autres événements.
Afin de démontrer que ce principes guide effectivement l’apprentissage biologique, l’équipe de Brice Bathellier (Institut des Neurosciences Paris Saclay) a utilisé deux méthodes optiques permettant de suivre et de modifier l’activité de larges ensembles de neurones définis génétiquement (optogénétique). Ils ont pu ainsi montrer que la quantité d’activité générée dans le système auditif corrèle avec la vitesse d’apprentissage lorsque des souris apprennent à associer des sons avec une récompense. Dans un deuxième temps ils ont pu faire apprendre aux souris à associer, non plus des sons, mais des activations précises, artificielles du système auditif. Grâce à cette manipulation, ils ont pu montrer un lien causal direct entre la quantité d’activité générée et la force du renforcement. Ainsi les neurones les plus actifs lors d’un événement sont bien ceux sélectionnés en priorité par les mécanismes d’apprentissage.
Référence
Ceballo et al., Cortical recruitment determines learning dynamics and strategy. Nature Communications, 10: 1479 (2019)
https://rdcu.be/budts
Contact Chercheur
Brice Bathellier
Paris-Saclay Institute of Neuroscience (NeuroPSI)
Department for Integrative and Computational Neuroscience (ICN)
UMR9197 CNRS/University Paris Sud
CNRS, Bldg. 32/33
1 Av. de la Terrasse, 91190 Gif-sur-Yvette, France
www.bathellier-lab.org
phone: +33 1 69823408
mail : bathellier@unic.cnrs-gif.fr
A key point of learning theory demonstrated through optogenetics
The brain learns constantly to improve our actions according to our experiences. Several theories aim to account for this fundamental property, one of the most popular being reinforcement learning, also used in artificial intelligence. This theory postulates that learning emerges through a specific reinforcement of connections between neurons that are active during an event, an action, or a sequence of events and actions leading to a reward. One of the key points of this theory is that the more active involved neurons are, the faster and more solid the reinforcement of connection is. Thus the events that most strongly activate our brain should be learned in priority with respect to other events.
In order to demonstrate that this principle actually guides biological learning, the team of Brice Bathellier (Paris Saclay Institute of Neuroscience) used two optical methods to read and modify the activity of large sets of genetically defined neurons (optogenetics). They were able to show that the amount of activity generated in the auditory system correlates with the speed of learning when mice learn to associate sounds with a reward. In a second experiment, they were able to teach mice to associate, no longer sounds, but precise, artificial activations of the auditory system. Through this manipulation, they were able to show a direct causal link between the amount of activity generated and the strength of reinforcement. Thus, the most active neurons during an event are those selected in priority by the learning mechanisms.
Des vagues corticales pour façonner la représentation du mouvement visuel
Comment le cerveau relie-t-il les informations visuelles dans l’espace et le temps ? Les illusions visuelles fournissent un paradigme expérimental pour étudier ces processus. Lorsque deux points sont présentés de manière statique et séquentielle à des positions différentes, l’observateur perçoit le mouvement d’un seul point allant d’une position à l’autre : le mouvement apparent. Pour de grandes séparations spatio-temporelles, le système visuel est mis au défi de relier ces informations et ainsi garder la trace de l’identité de l’objet le long du chemin du mouvement apparent, également connu sous le nom de “problème de correspondance”. Des chercheurs du CNRS (Marseille & Gif-sur-Yvette) et d’Aix-Marseille Université ont utilisé l’imagerie optique des colorants sensibles au potentiel de membrane dans le cortex visuel primaire (V1) de singes éveillés, combinée à la modélisation computationnelle, pour montrer que les connexions excitatrices et inhibitrices reliant les neurones séparés sur de longues distances à l’intérieur de V1, peuvent résoudre ce problème en liant les informations des deux stimuli dans l’espace et le temps. Ainsi deux vagues de propagation façonnent la représentation du mouvement illusoire, l’une facilitant la réponse dans la direction du mouvement, et l’autre, se déplaçant dans le sens opposé supprime la représentation résiduelle du premier stimulus. Les scientifiques proposent que cette vague suppressive soit un mécanisme de bas niveau pour résoudre les problèmes de correspondance ambigus et qu’elle contribuerait ainsi à encoder précisément la trajectoire du mouvement apparent à la surface de V1. Ces résultats, publiés dans The Journal of Neuroscience, démontrent quel rôle computationnel les vagues de propagation d’activité corticales peuvent jouer dans la représentation dynamique d’information sensorielle.
Référence
Chemla S, Reynaud A, di Volo M, Zerlaut Y, Perrinet L, Destexhe A & Chavane F. (2019).
Suppressive traveling waves shape representations of illusory motion in primary visual cortex of awake primate. Journal of Neuroscience, 2792-18.
Contact Chercheurs
Sandrine Chemla
Frédéric Chavane
Institut de Neurosciences de la Timone (INT), UMR 7289 CNRS & Aix-Marseille Université, Marseille
Courir ou manger du chocolat, un choix dicté par les récepteurs cannabinoïdes.

Les pathologies qui résultent de notre mode de vie sédentaire ont pour principale cause une inactivité physique, cette dernière étant souvent associée à une prise excessive de nourriture riche en sucres et/ou en gras. A l’opposé, une activité physique excessive aux dépens de la prise de nourriture peut également s’avérer nocive, comme l’illustrent des cas d’anorexie nerveuse. Ces données rendent donc cruciale la recherche des processus neurobiologiques contrôlant les motivations respectives pour l’activité physique et la prise alimentaire. Fruit de la collaboration entre des chercheurs de l’Inserm et du CNRS, une étude publiée le 07 Mars 2019 dans la revue JCI Insight révèle que les récepteurs cannabinoïdes CB1 jouent un rôle primordial dans le choix entre courir et consommer une nourriture chocolatée.
Les auteurs de ce travail avaient précédemment rapporté que les récepteurs des cannabinoïdes CB1, présents sur plusieurs types de neurones, jouent un rôle clef dans les performances lors d’une activité physique chez la souris. Cette conclusion était basée sur les performances réalisées par des animaux ayant un accès libre à une roue d’activité, un modèle qui ne permettait pas de distinguer le mécanisme mis en jeu (motivation, plaisir…). La motivation pour une récompense ne pouvant être estimée que par la mesure des efforts que l’individu, Homme ou animal, est prêt à fournir pour accéder à cette récompense, les chercheurs ont élaboré un modèle dans lequel chaque accès à la roue était conditionné par un effort préalable. Cet effort préalable consiste en l’introduction répétée du museau dans un réceptacle, condition sine qua none pour débloquer la roue. Après une période d’apprentissage de la tâche au cours de laquelle l’effort demandé était constant, les souris ont été confrontées à un test dans lequel l’effort demandé pour accéder à la roue a été augmenté de manière progressive. Exposées à ce test, des souris dépourvues de récepteurs CB1 ont montré un déficit de 80 % dans l’effort maximal qu’elles étaient prêtes à fournir pour accéder à la roue, et ce sans diminution des performances lors de leurs accès à la roue. Ce résultat indique que les récepteurs CB1 jouent un rôle majeur dans le contrôle de la motivation pour l’activité physique. L’utilisation d’autres souris génétiquement modifiées a également permis aux chercheurs de démontrer que ces récepteurs CB1 contrôlant la motivation pour l’exercice sont localisés sur des neurones GABAergiques.
Les chercheurs ont ensuite examiné si les récepteurs CB1 dans les neurones GABAergiques contrôlent la motivation pour une autre récompense, de la nourriture chocolatée (au même titre que les humains, les souris en raffolent même si elles sont bien nourries). Alors que les récepteurs CB1 jouent également un rôle dans la motivation pour la nourriture, mais à un degré moindre que dans la motivation pour l’activité physique, les récepteurs CB1 localisés sur les neurones GABAergiques ne sont pas impliqués dans la motivation pour la prise de nourriture chocolatée.
Dans notre vie quotidienne, nous sommes confrontés à un choix permanent entre plusieurs récompenses. Cette évidence a poussé les chercheurs à développer un modèle dans lequel, après apprentissage, les souris avaient le choix, moyennant les efforts décrits ci-dessus, entre une activité physique et de la nourriture chocolatée. La motivation pour l’activité physique l’a emporté sur la prise de nourriture chocolatée, à l’exception des souris dépourvues de récepteur CB1 de manière globale ou uniquement dans les neurones GABAergiques qui, elles, ont montré une préférence pour la nourriture.
Au-delà de ces résultats indiquant que le récepteur cannabinoïde est primordial pour la motivation pour l’activité physique, cette étude ouvre des perspectives pour pouvoir étudier les mécanismes neurobiologiques responsables d’augmentations pathologiques de cette motivation. Une illustration est fournie par l’anorexie nerveuse qui associe souvent une diminution de la motivation pour se nourrir à une augmentation de la motivation pour l’activité physique.
Source
The motivation for exercise over palatable food is dictated by cannabinoid type-1 receptors.
Muguruza C, Redon B, Fois GR, Hurel I, Scocard A, Nguyen C, Stevens C, Soria-Gomez E, Varilh M, Cannich A, Daniault J, Busquets-Garcia A, Pelliccia T, Caillé S, Georges F, Marsicano G, Chaouloff F.
JCI Insight. 2019 Mar 7;4(5). pii: 126190. doi: 10.1172/jci.insight.126190.
Contact chercheur
Francis Chaouloff
NeuroCentre INSERM U1215
Equipe “Endocannabinoïdes & NeuroAdaptation”
33077 Bordeaux
05 57 57 37 55
francis.chaouloff@inserm.fr
Des neurones de schéma dans le cerveau

Comment le cerveau se représente-t-il l’espace ? Des chercheurs du CNRS de Lyon et Grenoble ont observé l’activité cérébrale de macaques alors que ces animaux naviguaient dans des environnements 3D virtuels à la recherche d’une récompense. Ils ont montré que certains neurones d’une structure essentielle à la mémoire, l’hippocampe, permettent de mémoriser les détails des environnements (mémoire épisodique) tandis que d’autres encodent la logique d’organisation de l’espace, lorsqu’elle se répète. Ces « cellules de schéma » encodent le rôle fonctionnel attribué à des repères visuels plutôt que leur apparence. Ainsi, l’hippocampe pourrait représenter à la fois le caractère particulier d’expériences uniques en même temps que l’information commune à ces épisodes, réalisant un encodage compact des données à mémoriser. Ces résultats, publiés dans Science, montrent qu’une forme de pensée abstraite existe chez le macaque rhésus, et ouvrent ainsi des possibilités d’exploiter ce modèle animal pour faire progresser la compréhension de pathologies cliniques.
L’hippocampe est une structure du lobe temporal du cortex cérébral qui joue un rôle essentiel dans la mémoire des souvenirs (mémoire épisodique) ainsi que dans l’orientation spatiale. Des lésions de cette partie du cerveau, comme dans la maladie d’Alzheimer, mènent à des pertes mnésiques et de profondes désorientations. La collaboration entre des chercheurs de l’Institut des Sciences Cognitives Marc Jeannerod (Lyon, CNRS/Université de Lyon) et du GIPSA-lab (Grenoble, CNRS/UGA) a permis de préciser le rôle des neurones de l’hippocampe dans la mémoire spatiale chez le singe. Les animaux étaient plongés dans un environnement 3D virtuel (labyrinthe en étoile) où ils devaient trouver une récompense invisible en se repérant grâce à des éléments distants (des amers, p. ex. l’arbre de l’illustration). Après des semaines d’entraînement dans un environnement devenu familier, les animaux ont été testés dans des environnements à la géométrie identique mais dont les amers changent chaque jour. Quelques essais et erreurs sont alors suffisants aux animaux pour se repérer, démontrant ainsi leur compréhension de la tâche et de la structure de l’environnement : les animaux ont formé un schéma mental de l’environnement-type. Si beaucoup de neurones de l’hippocampe semblent coder des aspects uniques à chaque environnement, comme l’identité des amers, d’autres se comportent comme des “cellules de schéma” dont l’activité, une fois rapportée à la position de la récompense, est similaire dans tous les environnements. Ainsi, l’hippocampe pourrait représenter à la fois le caractère particulier d’expériences uniques en même temps que l’information commune à ces épisodes, réalisant un codage compact des données à mémoriser. Ceci est encore plus apparent si, au lieu de caractériser l’activité des neurones par rapport à la position de l’animal dans l’espace physique, on la repère dans un espace des états de la tâche, une représentation plus abstraite de la progression de l’animal vers son but, à l’origine utilisée en automatique ou en robotique, et prenant en compte la position, l’orientation et l’historique de navigation de l’animal. Les neurones de schéma utilisent donc tous ces indices pour construire une représentation fonctionnelle de l’environnement. Ce modèle animal pourrait être utile dans la compréhension de certaines pathologies cliniques.
Référence
Baraduc P, Duhamel JR, Wirth S (2019). Schema cells in the macaque hippocampus. Science, 2019 Feb 8; 363(6427):635-639.
Contact chercheurs:
Pierre Baraduc, GIPSA-lab, CNRS/U.Grenoble-Alpes, 11 rue des Mathématiques, 38402 Saint Martin d’Hères. 04 76 82 71 50. pierre.baraduc@gipsa-lab.fr
Sylvia Wirth, ISCMJ, CNRS/U.Lyon, 67 Bd Pinel, 69675 Bron. 04 37 91 12 32. sylvia.wirth@isc.cnrs.fr
La preuve est faite : Les champs magnétiques des structures profondes du cerveau sont visibles depuis la surface !
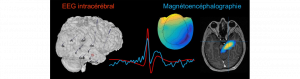
Une équipe pluridisciplinaire formée d’ingénieurs et de cliniciens (AMU, Inserm, AP-HM) vient de faire la démonstration qu’il est possible de détecter en surface des activités pathologiques se produisant dans des structures profondes du cerveau (Pizzo et al. Nat Comm 2018). Ces structures sont fortement impliquées dans des pathologies comme l’épilepsie ou certaines maladies neuro-dégénératives. Elles étaient considérées jusqu’à présent comme invisibles à partir de la surface, nécessitant l’implantation d’électrodes directement dans le cerveau par la technique d’EEG intracérébrale stéréotaxique (SEEG). Les chercheurs ont utilisé une combinaison unique d’enregistrements simultanés de magnétoencéphalographie (MEG) et de stéréo-électroencéphalographie (SEEG), et des méthodes avancées de traitement du signal. Ces résultats ouvrent de nouvelles possibilités dans l’étude non-invasive de la dynamique cérébrale, à la fois en clinique et en neurosciences fondamentales.
La MEG est une technique de pointe non-invasive utilisée pour cartographier les activités cérébrales, qui possède une excellente résolution à la fois spatiale et temporelle. La SEEG est une technique invasive utilisée lors du bilan préchirurgical des patients épileptiques, consistant à implanter des électrodes directement dans le cerveau. Très peu de centres au niveau mondial maîtrisent l’enregistrement simultané de ces deux méthodes, une prouesse technique qui a été rendue possible grâce à une collaboration rapprochée entre recherche et clinique. Ces enregistrements simultanés ont permis de confirmer la capacité de la MEG d’enregistrer le signal des zones du cerveau. Cela ouvre à terme la possibilité pour certains patients de se passer d’enregistrement invasifs, ce qui serait une grande avancée.
Les chercheurs et enseignants-chercheurs de l’Institut de Neurosciences des Systèmes (INS, Aix-Marseille Université et Inserm) et du service d’Epileptologie et de Rythmologie Cérébrale de l’assistance publique des hôpitaux de Marseille (AP-HM) ont ainsi pu montrer que des activités enregistrées avec des électrodes profondes dans l’hippocampe, l’amygdale et le thalamus produisent bien un reflet mesurable en surface sur les capteurs de MEG. Cela résout une controverse existante de longue date, car il est communément admis que des structures cérébrales aussi profondes et d’architecture complexe ne sont pas visibles directement, mais plutôt indirectement par propagation neuronale vers des structures plus superficielles. Grâce au traitement du signal, les chercheurs ont pu séparer les deux types d’activités, propagée et initiale, et ainsi démontrer que cette dernière est bien visible en surface.
Ces structures profondes du cerveau (en particulier du lobe temporal) sont impliquées à la fois dans le fonctionnement normal (mémoire, émotions) et dans le dysfonctionnement (épilepsie, maladies neurodégénératives) du cerveau. Cette découverte a donc des conséquences à la fois au niveau clinique, car elle suggère que l’on peut se passer d’implanter des électrodes pour diagnostiquer le cerveau, et au niveau des neurosciences, car elle ouvre la voie à de nouvelles études sur la dynamique spatio-temporelle des réseaux cérébraux.
Reference
Pizzo F, Roehri N, Medina Villalon S, Trébuchon A, Chen S, Lagarde S, Carron R, Gavaret M, Giusiano B, McGonigal A, Bartolomei F, Badier JM, Bénar CG. Deep brain activities can be detected with magnetoencephalography. Nat Commun. 2019 Feb 27;10(1):971. doi: 10.1038/s41467-019-08665-5.
Contact chercheur
Christian Bénar
Aix Marseille Univ, INSERM, INS, Inst Neurosci Syst, Marseille, 13005, France.
christian.benar@univ-amu.fr.
Trois neurotransmetteurs et la maladie de Parkinson
La perte de l’innervation dopaminergique des ganglions de la base et notamment du striatum dorsal (putamen) est à l’origine des signes moteurs de la maladie de Parkinson : une lenteur à exécuter les mouvements, une rigidité musculaire et des problèmes de posture. Si la quasi-totalité des neurones du striatum sont GABAergiques, seulement1% sont des interneurones cholinergiques. Ces interneurones géants génèrent une réponse caractéristique à un stimulus sensoriel associé à une récompense, une pause dans leur décharge, qui disparait dans le striatum privé de ses afférences dopaminergiques.
Un troisième neurotransmetteur,l’acide g-aminobutyrique (GABA), s’est invité dans la pathologie lorsque nous avons découvert qu’environ 50% des interneurones cholinergiques du striatum sont aussi GABAergiques, en nous basant sur des marqueurs (Lhx6), de la PCR intracellulaire (gad2) et des enregistrements de paires de neurones. Faiblement connectés entre eux chez les animaux témoins,ces interneurones mixtes libèrent acétylcholine et GABA, agissant sur des récepteurs nicotiniques et GABAA, pour générer des courants antagonistes, entrant et sortant, respectivement. Dans le striatum déplété endopamine, les connexions entre neurones mixtes augmentent et le courant GABAA devient entrant, tout comme le courant cholinergique, abolissant ainsi la« pause-réponse » évoquée par la stimulation des afférences corticales. Baisser les taux de (Cl–)i avec la bumétanide, un bloquant de l’importateur des ions chlorures, NKCC1, rétablit la polarité du courant GABAA et la pause-réponse in vitro. In vivo, le traitement chronique à la bumétanide des souris dont un des striatum est déplété en dopamine, améliore leur motricité.
En parallèle, nous avons conduit un essai clinique ouvert sur 4 patients avec des effets prometteurs de la bumétanide. Nous allons bientôt effectuer un essai en double aveugle sur 40patients, centré sur les problèmes de chute et de « freezing » des parkinsoniens. Collectivement, ces travaux illustrent l’importance du métabolisme neuronal des ions chlorures, perturbé dans de nombreuses pathologies neurologiques.
Three neurotransmitters and Parkinson disease
The progressive loss of dopaminergic innervation of the basal ganglia, in particular of the dorsal striatum (putamen) is responsible for the motor signs of Parkinson disease such as bradykinesia, rigidity and loss of postural reflexes. In the striatum, the vast majority of neurons are GABAergic, with cholinergic interneurons representing only 1% of the total population. These giant interneurons gradually develop a typical conditioned pause response during acquisition of a sensorimotor association. The proportion of these responsive interneurons is dramatically decreased in the dopamine-depleted striatum.
We recently identified a third player in the disease, the gamma-aminobutyric acid (GABA). On the basis of intracellular markers labeling (Lhx6), RTqPCR (gad2) and pair recordings, we showed that around 50% of cholinergic interneurons also synthesize and release GABA. Loosely interconnected in control striatum, these mixed interneurons generate inward nicotinic and outward GABAA currents. In the dopamine-depleted striatum, interconnections dramatically increase and the GABAA current becomes inward like the nicotinic one, leading to disappearance of the pause (suppression of firing) in response to cortical afferents stimulation. Application in slices of the blocker of the chloride importer NKCC1, bumetanide, to lower intracellular chloride concentration, reestablishes GABAAcurrent polarity and the pause response. Chronic bumetanide treatment ameliorates the motricity of mice with dopamine-depletion in one striatum.
In parallel, we conducted a promising open study with 4 patients which will be followed by a double-blind essay with 40 patients, targeting problems of posture, falls and freezing, in particular. Overall our studies illustrate the importance of neuronal chloride metabolism which is altered in a variety of neurological disorders.
Référence
GABAergic inhibition indual-transmission cholinergic and GABAergic striatal interneurons is abolishedin Parkinson disease. LozovayaN, Eftekhari S, Cloarec R, Gouty-Colomer LA, Dufour A, Riffault B, Billon-Grand M, Pons-Bennaceur A, Oumar N, Burnashev N, Ben-Ari Y, Hammond C.
Nat Commun. 2018 Apr 12;9(1):1422. doi: 10.1038/s41467-018-03802-y.
Contact chercheur
Constance Hammond
Inmed Inserm UMR 1249 et B&A Therapeutics, 163 avenue de Luminy, Marseille.
constance.hammond@inserm.fr
Les séquences neurales enchevêtrées sont indispensables à la formation de la mémoire !

De nombreuses fonctions cognitives semblent être sous-tendues au niveau cérébral par la formation de séquences d’activité neuronale, c’est-à-dire par l’activation successive d’ensembles spécifiques de neurones, dans un ordre bien précis. Ces fonctions sont variées, allant de la vocalisation chez les oiseaux, à la réactivation de souvenirs chez les primates, en passant par la discrimination d’odeurs chez les criquets, ou encore la planification et la prise de décision chez les rats. Les séquences d’activité peuvent se produire plus ou moins rapidement, depuis l’échelle de temps lente du comportement (c’est-à-dire conditionnée par la perception ou par l’action), jusqu’à l’échelle de temps rapide endogène (c’est-à-dire conditionnée par les propriétés intrinsèques des réseaux de neurones concernés). Un exemple particulièrement frappant est l’hippocampe, dont les cellules de lieu codent la position de l’animal dans l’environnement. Lorsque l’animal se déplace, les cellules de lieu s’activent les unes après les autres au fil de la trajectoire, ce qui forme des séquences d’activité à l’échelle de temps comportementale. Ensuite, pendant le sommeil, ces mêmes séquences se reproduisent spontanément, comme si l’animal « rêvait » des trajectoires qu’il vient de parcourir. Ces réactivations sont hautement accélérées, environs vingt fois plus rapides, et permettent de renforcer la mémoire pendant le sommeil. Comment l’organisation séquentielle des cellules de lieu peut-elle être maintenue à des échelles de temps si différentes, manifestées à des moments totalement séparés dans le temps, et dans des états cérébraux opposés (veille, sommeil) ?
Une première possibilité est que l’information séquentielle est directement enregistrée lorsque les neurones s’activent les uns après les autres à l’échelle de temps du comportement. Une seconde possibilité, plus énigmatique, met en jeu la remarquable propriété de l’hippocampe de générer des séquences d’activité enchevêtrées, c’est-à-dire mêlant de manière profondément intriquée des séquences lentes et des séquences rapides. Ceci se produit en fait pendant l’exploration : alors même que les cellules de lieu s’activent lentement les unes après les autres, le réseau hippocampique produit également des séquences d’activité à l’échelle de temps d’une oscillation cérébrale appelée « thêta », dont les cycles durent à peine 150 ms. Ceci permet aux cellules de lieu de s’activer de manière répétée, très rapidement les unes après les autres, comme si à chaque instant elles représentaient toute la trajectoire en cours. Grâce à leur vitesse élevée, ces séquences enchevêtrées permettraient aux cellules de lieu de renforcer leurs connexions, et ainsi de mémoriser leur séquence d’activation. Mais s’agit-il vraiment là du mécanisme qui permet à l’hippocampe de mémoriser des trajectoires, ou d’un simple épiphénomène, si frappant soit-il ?
Pour répondre à cette question, nous avons enregistré des séquences d’activité hippocampiques chez des rats, pendant l’exploration de l’environnement, et pendant le sommeil. Nous avons développé un protocole, grâce auquel nous avons pu perturber, de manière rapide et sélective, les séquences d’activité enchevêtrées, sans toucher aux séquences lentes. Ainsi, les cellules de lieu s’activaient-elles toujours les unes après les autres à mesure que les rats parcouraient l’environnement (à l’échelle de temps du comportement), mais les séquences enchevêtrées (à l’échelle de temps du rythme thêta) pouvaient être supprimées à volonté. Nous avons constaté que la perturbation des séquences enchevêtrées avait pour conséquence une absence totale de réactivations pendant la période de sommeil suivante, réactivations qui permettent normalement la consolidation de la mémoire. Ainsi est-ce bien grâce à son étonnante capacité à produire en même temps des séquences rapides et lentes, grâce à cet enchevêtrement des échelles de temps, que l’hippocampe peut mettre initialement en mémoire les souvenirs qui seront ensuite renforcés pendant le sommeil pour permettre une mémorisation à long terme.
Référence
Drieu C, Todorova R, Zugaro M.. Nested sequences of hippocampal assemblies during behavior support subsequent sleep replay. Science. 2018 Nov 9;362(6415):675-679. doi: 10.1126/science.aat2952.
Contact chercheur
Center for Interdisciplinary Research in Biology (CIRB), Collège de France, CNRS, INSERM, PSL Research University, Paris.
L’acétylcholine et les circuits neuronaux de la dépression
Selon l’Organisation Mondiale de la Santé, la dépression est un trouble mental courant affectant plus de 300 millions de personne dans le monde. L’apparition de troubles dépressifs est la conséquence d’une interaction complexe entre prédispositions génétiques et facteurs psychosociaux. Elle se caractérise notamment par une perte de plaisir (anhédonie) et l’évitement des autres (aversion sociale). Ces altérations comportementales peuvent être observées chez des souris soumises à des expériences traumatogènes comme un stress social. En effet, l’exposition répétée de souris à des congénères mâles dominants entraine l’apparition de troubles comportementaux caractéristiques de la dépression associés à la dérégulation d’un messager chimique du cerveau, la dopamine. Comprendre les mécanismes impliqués dans ces adaptations est un enjeu important pour le traitement des troubles psychiatriques liés au stress.
Les récents travaux de notre équipe au sein de l’Institut de Pharmacologie Moléculaire et Cellulaire, publiés dans la revue Nature Communications, ont dévoilé en partie les mécanismes neurobiologiques impliqués dans les troubles dépressifs. Nous avons identifié un circuit neuronal qui sous-tend la dérégulation des signaux dopaminergiques et l’apparition des comportements anhédoniques et d’aversion sociale. En effet, nous avons montré que l’exposition à un stress social chronique entraine une profonde dérégulation des neurones du noyau latérodorsal du tegmentum (LDTg), notamment ceux produisant l’acétylcholine, un messager chimique jouant un rôle important dans la modulation de l’activité des neurones dopaminergiques. Le blocage sélectif de l’activité de ces neurones par l’utilisation d’approches chimiogénétiques lors des phases d’exposition au stress est suffisante pour empêcher la cascade de réactions aboutissant à l’apparition des troubles comportementaux. La dérégulation des neurones à acétylcholine est causée par le relargage de corticolibérine et sa fixation sur le récepteur CRF de type 1 modulant directement l’activité des neurones cholinergiques.
Ces résultats pourraient mener à de nouvelles pistes thérapeutiques pour combattre la dépression, soit par des approches pharmacologiques contre les cibles moléculaires identifiées, soit par des approches permettant de moduler l’activité des macrocircuits mis en évidence.
Reference
Fernandez SP, Broussot L, Marti F, Contesse T, Mouska X, Soiza-Reilly M, Marie H, Faure P, Barik J.
Mesopontine cholinergic inputs to midbrain dopamine neurons drive stress-induced depressive-like behaviors.
Nat Commun. 2018 Oct 25;9(1):4449. doi: 10.1038/s41467-018-06809-7.
Contact chercheur
Jacques Barik
Université Côte d’Azur, Nice, 06560, France
Institut de Pharmacologie Moléculaire & Cellulaire, CNRS, UMR7275, Valbonne, France
barik@ipmc.cnrs.fr
Estimates from the WHO show that depression is one of the most common psychiatric disorders, affecting 300 million people worldwide. Depression appears to be the result of a complex interaction between genetic predisposition and psychosocial factors. Most common symptoms include decrease ability to experience pleasure (anhedonia) and strong withdrawal from social interaction. These behavioral traits can be modeled and studied in mice subjected to stressful events such as social defeat. Indeed, in mice repeated exposure to a dominant conspecific produces depressive-like behavioral alterations that have been associated with an imbalance in the brain neurotransmitter dopamine. Understanding the process underlying these mal-adaptations warrants better treatments for psychiatric disorders associated with stress.
In a recent work published by our team (Institute of Molecular and Cellular Pharmacology) in the journal Nature Communications, we unraveled a novel mechanism implicated in the neurobiology of depression. We identified a neuronal circuit that promotes a dysregulation in dopaminergic signaling and the appearance of social aversion and anhedonia, two hallmarks of depressive states. We showed that exposure to chronic social stress induces strong activation of cholinergic neurons in the laterodorsal tegmental (LDTg) nucleus, which are important regulators of dopamine neurons activity. Selective chemogenetic silencing of these cholinergic neurons during social stress exposure was sufficient to prevent the chain of events linked with maldaptive behavioral responses. The dysregulation of LDTg cholinergic neurons by stress involves the release of corticotrophin-releasing factor (CRF) and selective activation of CRF-1 receptors in these neurons.
These results will open up new avenues in the fight against depression, by pinpointing a molecular target for drug discovery, or alternatively by brain neuromodulation approaches on these newly described circuit mechanisms.