Syndrome de Rett : Lorsque la huntingtine sauve les neurones déficients par Contributeur 31.01.2020 à 02h35
Une étude récente menée entre autres à Aix-Marseille Université et l’Inserm laisse entrevoir un espoir thérapeutique dans le syndrome de Rett ! En effet, les équipes de Jean-Christophe Roux (équipe de Neurogénétique Humaine) du Centre de Génétique Médicale de Marseille* et Frédéric Saudou du Grenoble Institut Neurosciences** ont élaboré une stratégie originale en axant leurs travaux sur la huntingtine, protéine impliquée dans la maladie de Huntington.
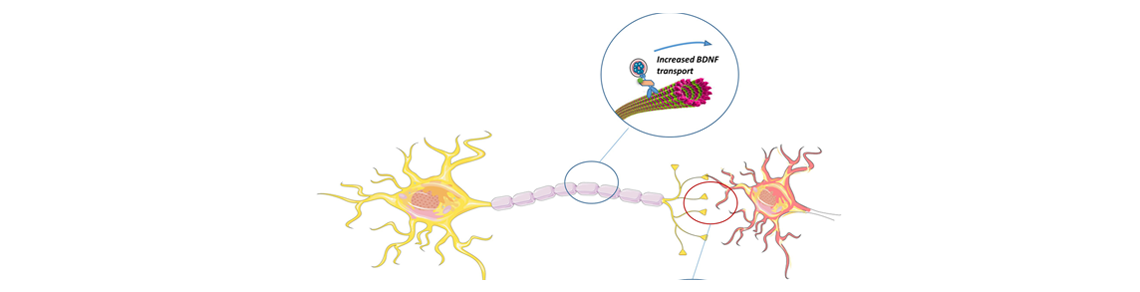
Le syndrome de Rett est un trouble neurologique grave qui affecte uniquement des filles avec une incidence de 1 sur 15 000 naissances. Ce n’est qu’entre 6 à 18 mois que les premiers signes apparaissent. Pour l’instant, il n’existe aucun traitement pour cette pathologie qui conduit à un polyhandicap sévère se traduisant par des troubles cognitifs, moteurs et autonomes. L’activation de la protéine Huntingtine, qui lorsque mutée est responsable de la maladie de Huntington (MH), améliore la physiopathologie et les symptômes dans des souris modèles du syndrome de Rett. En effet, l’élévation de son taux de phosphorylation permet de restaurer le transport endogène de BDNF – facteur neurotrophique dérivé du cerveau – essentiel au développement et au bon fonctionnement des neurones. De nombreuses études ont ainsi établi un lien direct entre la réduction des connexions neuronales et l’altération du taux de sécrétion du BDNF. Chez les malades atteints du syndrome de Rett, le gène responsable a été identifié en 1999. Il s’agit de MECP2 qui régule l’expression de milliers de gènes neuronaux dont celle du BDNF. Lorsque MCP2 est muté, l’expression du BDNF est diminuée de moitié, provoquant de nombreux problèmes de connectivité neuronale. En utilisant des approches génétiques et pharmacologiques, Jean-Christophe Roux et Frédéric Saudou ont montré que la phosphorylation de la huntingtine permet de pallier ce problème en augmentant le transport de BDNF dans des neurones modèles de Rett et améliore ainsi le fonctionnement des synapses. Des travaux antérieurs avaient identifié la molécule FK506, déjà utilisée en clinique pour éviter le rejet de greffe, comme augmentant la phosphorylation de la huntingtine. Les chercheurs ont alors montré que cette molécule était capable de rétablir le transport de BDNF dans les puces microfluidiques reproduisant les connexions dans les cerveaux Rett et améliorait la physiopathologie et les symptômes chez les souris modèle du syndrome de Rett.
Ces travaux, publiés dans le journal Embo Molecular Medicine le 08/01/2020, pourraient constituer une approche prometteuse pour le traitement des patientes atteintes du syndrome de Rett.
Référence : Ehinger Y, Bruyère J, Panayotis N, Abada YS, Borloz E, Matagne V, Scaramuzzino C, Vitet H, Delatour B, Saidi L, Villard L, Saudou F, Roux JC. Huntingtin phosphorylation governs BDNF homeostasis and improves the phenotype of Mecp2 knockout mice. EMBO Mol Med. 2020 Jan 8:e10889. doi: 10.15252/emmm.201910889.
Contact chercheur :
Jean-Christophe Roux, Équipe de NeurogénétiqueHumaine, MMG – Centre de génétique médicale de Marseille, Aix-Marseille Université – Inserm – UMR 1251, Faculté de Médecine de La Timone, Marseille.
Frédéric Saudou, Directeur du Grenoble institut des Neurosciences (GIN), Équipe “Dynamiques intracellulaires et neurodégénérescence”, Inserm, U1216, CHU Grenoble Alpes, Grenoble