Maladie de Huntington: des anomalies cérébrales détectables dès le stade embryonnaire chez l’homme par Contributeur 14.10.2020 à 10h55
La maladie de Huntington – comme la maladie d’Alzheimer, de Parkinson et la sclérose latérale amyotrophique- fait partie de la famille des maladies qui se manifestent à l’âge adulte alors que -en ce qui concerne les formes héréditaires- l’anomalie génétique est présente dès la conception. Par exemple, la protéine huntingtine (HTT) dont la mutation du gène par expansion de triplets CAG conduit à la maladie, est exprimée très précocement dans le développement pendant lequel elle joue un rôle essentiel. La HTT mutante interfère avec plusieurs étapes du développement de certaines régions cérébrales, dont le cortex. De plus, l’expression de la HTT mutante restreinte au développement suffit à produire des caractéristiques de la maladie de Huntington chez des souris adultes suggérant qu’il y a une composante développementale à la maladie.
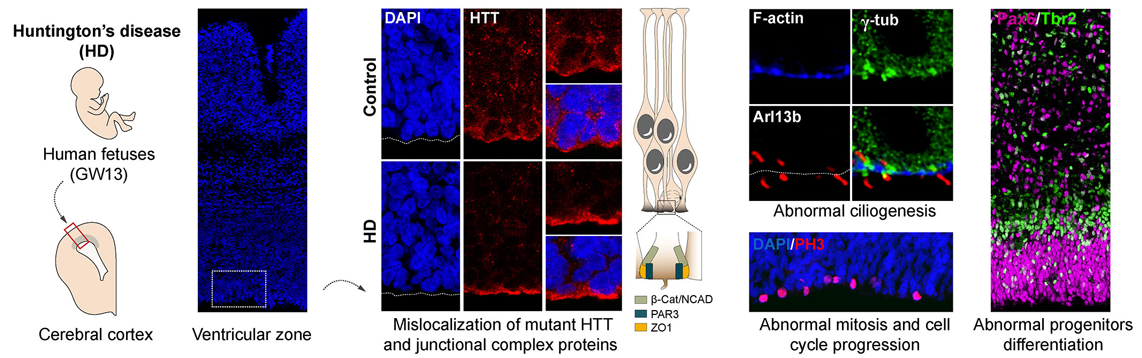
Savoir si le développement cérébral humain précoce est modifié restait une question centrale dans le domaine. Pour y répondre, les équipes de Sandrine Humbert, directrice de recherche Inserm au Grenoble Institut des neurosciences (Inserm/Université Grenoble Alpes), et Alexandra Durr, professeur des universités-praticien hospitalier à Sorbonne Université, à l’Hôpital de la Pitié Salpêtrière – AP-HP et à l’Institut du cerveau (Inserm/Sorbonne Université/CNRS/AP-HP) ont examiné des tissus de fœtus humains porteurs de l’expansion pathologique de triplet CAG dans le gène HTT, issus de dons des parents suite à une interruption médicale de grossesse. Ces tissus présentent des anomalies dans le cortex en développement, notamment une mauvaise localisation de la huntingtine mutante et de protéines de complexes de jonction, des défauts de polarité et de différenciation des précurseurs neuronaux, une ciliogénèse anormale et des changements dans la mitose et la progression du cycle cellulaire. Ces anomalies perturbent l’équilibre « division-différentiation » des cellules progénitrices. Celles-ci sont en effet issues d’un réservoir de cellules en division dont une partie se différencie en neurones tandis que l’autre continue de se diviser pour fournir de nouvelles cellules progénitrices. Chez les embryons porteurs de la mutation, ces cellules progénitrices entrent plus vite en différenciation au dépend du réservoir de cellules en division.
Ces travaux fournissent donc la première preuve directe provenant de fœtus humains que le développement du cerveau est altéré dans une maladie neurodégénérative se manifestant à l’âge adulte. Cette découverte ouvre un champ de nouvelles investigations visant à comprendre comment ces défauts précoces contribuent à la pathologie adulte et comment la compensation de ces derniers est régulée. Cette découverte a aussi des conséquences importantes pour la façon et le stade auxquels les traitements qui modifient le cours de la maladie doivent être envisagés.
Reference
Huntington disease alters human neurodevelopment. M Barnat, M Capizzi, E Aparicio, S Boluda, D Wennagel, R Kacher, R Kassem, S Lenoir, F Agasse, BY Braz, JP Liu, J Ighil, A Tessier, SO Zeitlin, C Duyckaerts, M Dommergues, A Durr, S Humbert. Science, 2020, 10.1126/science.aax3338
Contact chercheuses
Univ.Grenoble Alpes, INSERM,U1216, Grenoble Institut Neurosciences, Grenoble, France
Institut du Cerveau et de la MoelleEpinière (ICM), Genetics, AP HP, Sorbonne University, InsermU1127, CNRSUMR7225, Pitié-Salpêtrière Hospital, Paris, France.
Huntington disease alters human neurodevelopment
Huntington disease – like amyotrophic lateral sclerosis and Alzheimer’s and Parkinson’s disease – is part of the family of diseases that shared a delayed onset in mid-adulthood despite the expression, at least in hereditary cases, of the disease-driving protein from the first days of life. For example, huntingtin, the protein mutated in Huntington disease, is expressed very early in development during which it plays a key role. In mice, mutant huntingtin interferes with several stages of development in certain brain regions, including the cortex. Furthermore, expression of mutant huntingtin restricted to development is sufficient to produce features of Huntington’s disease in adult mice suggesting that there is a developmental component to the disease.
Nevertheless, whether early human brain development is altered remained a central question in the field. To answer it, teams led Sandrine Humbert, research director, INSERM (the French National Institute for Health and Medical Research) and group leader at the Grenoble Institut des Neurosciences, and by Alexandra Durr, professor at Sorbonne University and team leader in the Paris Brain Institute at Pitie-Salpêtrière Hospital, Paris had access to fetal tissue from families that terminated their pregnancy in the context of a prenatal test. The developing fetus carried the Huntington disease gene mutation. These tissues showed abnormalities in the developing cortex, including abnormal localization of mutant huntingtin and junction complex proteins, defects in polarity and differentiation of neural precursors, abnormal ciliogenesis, and changes in mitosis and cell cycle progression. These abnormalities disrupt the « division-differentiation » balance of progenitor cells. Progenitor cells come from a pool of dividing cells, some of which differentiate into neurons while the other continues to divide to provide new progenitor cells. In Huntington disease gene carrier embryos, these progenitor cells differentiate more rapidly at the expense of the pool of dividing cells.
This work provides the first direct evidence from human fetuses that brain development is impaired in a neurodegenerative disease with delayed onset. This discovery opens up a field of new investigations aimed at understanding how these early defects contribute to adult pathology and how their compensation is regulated. This discovery also has important implications for the way and stage at which disease-modifying treatments should be considered.